Apoptosis – a. Definition of Apoptosis b. Enumerate in Detail about the Pathways of Apoptosis c. What are the Other Methods of Cell Death? Answer the following question has 2nd yr mbbs student who is appearing for theory exam
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have all the information needed for a comprehensive, exam-ready answer. Let me compile it.
APOPTOSIS - Complete Answer for 2nd Year MBBS Theory Exam
(a) Definition of Apoptosis
Apoptosis (Greek: "falling off," like petals from flowers) is a pathway of programmed cell death in which cells activate enzymes - specifically cysteine proteases called caspases - that degrade the cell's own nuclear DNA and nuclear and cytoplasmic proteins.
Key defining features:
- The plasma membrane remains intact throughout the process
- The cell breaks into membrane-bound fragments called apoptotic bodies
- These are rapidly phagocytosed by macrophages without triggering inflammation
- The cell "dies with dignity" - its contents never leak into the surrounding tissue
Apoptosis is distinct from necrosis, where cell death is accidental, accompanied by membrane rupture, and causes an inflammatory reaction in surrounding tissue.
| Feature | Apoptosis | Necrosis |
|---|---|---|
| Cell size | Shrinkage | Swelling |
| Plasma membrane | Intact | Disrupted |
| Nucleus | Fragmented (karyorrhexis) | Karyolysis / pyknosis |
| Inflammation | Absent | Present |
| DNA fragmentation | Ladder pattern (nucleosomal) | Random |
| Caspase activation | Yes | No |
| Trigger | Programmed / physiological | Pathological (injury) |
(Robbins & Kumar Basic Pathology; Histology - A Text and Atlas)
Causes of Apoptosis
Physiological:
- Embryogenesis (tissue patterning, digit formation)
- Turnover of proliferative tissues (intestinal epithelium, lymphocytes)
- Involution of hormone-dependent tissues (endometrium after menstrual cycle)
- Elimination of self-reactive lymphocytes (prevents autoimmunity)
- Decline of leukocytes after an immune/inflammatory response
Pathological:
- DNA damage (radiation, cytotoxic drugs)
- Accumulation of misfolded proteins (ER stress)
- Certain viral infections
- Severe cellular stress (hypoxia, toxins)
(b) Pathways of Apoptosis
Both pathways converge on the activation of caspases - ultimately the executioner caspases (caspase-3 and caspase-7).
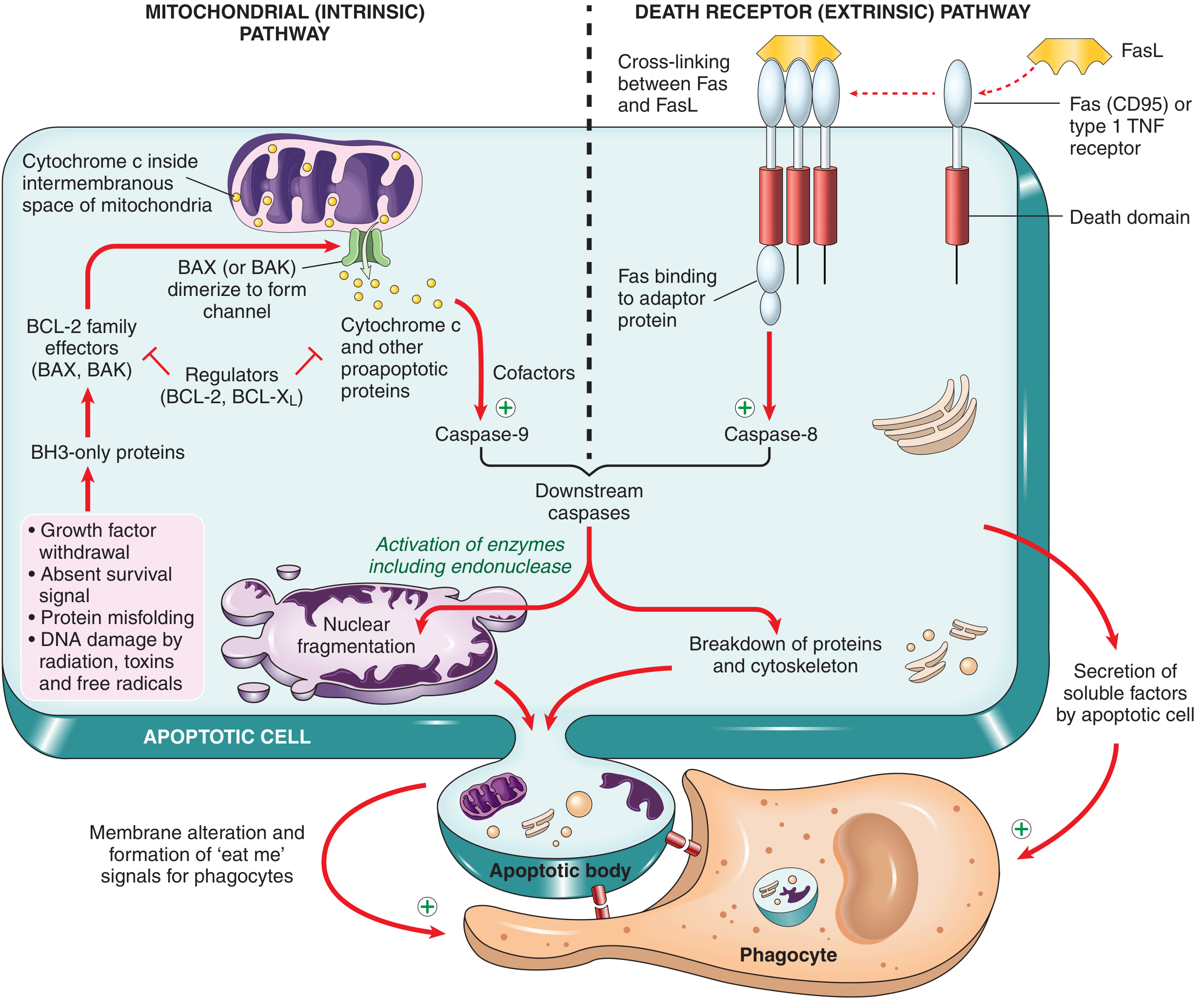
PATHWAY 1: Mitochondrial (Intrinsic) Pathway
This is the most common pathway, responsible for most physiologic and pathologic apoptosis.
Trigger stimuli:
- Loss of growth factors / survival signals
- DNA damage (radiation, free radicals, toxins)
- Accumulation of misfolded proteins (ER stress)
- Protein misfolding
Key molecules - the BCL-2 family:
The BCL-2 family of proteins controls the permeability of mitochondrial membranes. They fall into 3 groups:
| Group | Members | Function |
|---|---|---|
| Antiapoptotic | BCL-2, BCL-XL | Maintain mitochondrial integrity; keep BAX/BAK in check |
| Proapoptotic effectors | BAX, BAK | Dimerize to form channels in outer mitochondrial membrane |
| BH3-only sensors | BID, BAD, NOXA, PUMA | Sense cellular stress; shift balance toward BAX/BAK |
Sequence of events:
- Cell stress activates BH3-only proteins (sensors)
- BH3-only proteins inhibit BCL-2/BCL-XL, while activating BAX and BAK
- BAX and BAK dimerize and insert into the outer mitochondrial membrane, forming channels
- Cytochrome c (and other proapoptotic proteins like SMAC/DIABLO) leak into the cytosol
- In the cytosol, cytochrome c binds with Apaf-1 (apoptotic protease activating factor-1) to form the apoptosome
- The apoptosome activates caspase-9 (initiator caspase)
- Caspase-9 activates caspase-3 and caspase-7 (executioner caspases)
- Executioner caspases cleave structural proteins, activate endonucleases -> nuclear fragmentation
Memory tip: BCL-2 = survival; BAX/BAK = death; BH3 = sensor/amplifier
PATHWAY 2: Death Receptor (Extrinsic) Pathway
This pathway is triggered by external "death signals" via specific surface receptors.
Trigger stimuli:
- Fas ligand (FasL) expressed on activated cytotoxic T lymphocytes
- Tumor necrosis factor (TNF)
- TRAIL (TNF-related apoptosis-inducing ligand)
Key receptors:
- Fas (CD95) - prototype death receptor
- Type I TNF receptor (TNFR1)
These receptors contain a cytoplasmic "death domain" - a conserved region that mediates interaction with downstream signaling proteins.
Sequence of events:
- FasL (on T lymphocyte) binds Fas (CD95) on the target cell
- Fas molecules are cross-linked, and the death domain recruits adaptor proteins (e.g., FADD - Fas-associated death domain)
- These adaptor proteins recruit and activate caspase-8 (initiator caspase)
- This forms the DISC (Death-Inducing Signaling Complex)
- Caspase-8 directly activates executioner caspases (caspase-3 and 7)
- Nuclear fragmentation, cytoskeletal breakdown, and apoptotic body formation
The extrinsic pathway is particularly important for:
- Elimination of self-reactive lymphocytes (immune tolerance)
- Killing of virus-infected or tumor cells by cytotoxic T lymphocytes
Cross-talk Between the Two Pathways
Caspase-8 (activated by the extrinsic pathway) can cleave BID (a BH3-only protein), converting it to truncated BID (tBID), which then activates the mitochondrial pathway. This amplifies the apoptotic signal, especially in cells where the extrinsic pathway alone is insufficient.
Terminal Phase (Common to Both Pathways)
Once either caspase-8 or caspase-9 is activated:
- Downstream executioner caspases (3 and 7) are activated
- These activate endonucleases -> DNA fragmentation into oligonucleosomal pieces (characteristic "ladder" pattern on gel electrophoresis)
- Cytoskeletal breakdown and nuclear condensation/fragmentation (karyorrhexis)
- Membrane blebbing and formation of apoptotic bodies
Clearance of Apoptotic Cells
- Phosphatidylserine (normally on the inner leaflet) flips to the outer leaflet of the plasma membrane - this is an "eat-me" signal
- Apoptotic cells secrete chemotactic signals that recruit macrophages
- Macrophages phagocytose apoptotic bodies rapidly and without inflammation
Third Pathway: Cytotoxic T Lymphocyte (CTL) - Mediated Pathway
CTLs can also kill target cells by secreting:
- Perforin - creates pores in the target cell membrane
- Granzymes - serine proteases that enter through the pores and activate caspase-3 directly
This mechanism combines features of both apoptosis (caspase activation) and necrosis (membrane poration). (Histology - A Text and Atlas)
(c) Other Methods of Cell Death
Beyond classic apoptosis and necrosis, several programmed non-apoptotic cell death modalities exist, mostly characterized by membrane rupture and caspase independence:
1. Necroptosis
- Features of both necrosis and apoptosis - "regulated necrosis"
- Initiated by TNF receptors and Fas signaling (similar to the extrinsic pathway), BUT proceeds via a caspase-independent mechanism
- Morphologically looks like necrosis (cell swelling, membrane rupture)
- Mediated by RIP1 and RIP3 kinases (receptor-interacting proteins)
- Inhibited by Necrostatin-1 (a specific inhibitor)
- Significance: involved in ischemic tissue damage
2. Pyroptosis ("Pyro" = fire/fever)
- Form of cell death induced by infection with certain microorganisms
- Dependent on caspase-1 (not involved in apoptosis)
- Inflammasome activation -> caspase-1 cleavage
- Releases proinflammatory cytokines IL-1β and IL-18 -> intense inflammation and fever
- Results in cell lysis and release of DAMPs (danger-associated molecular patterns)
- Important in the innate immune defense against intracellular pathogens
3. Autophagy ("Self-eating")
- Regulated cellular process for recycling cellular components via lysosomal degradation
- Process: intracellular membrane (from sER) wraps around organelle/cytoplasm -> autophagosome -> fuses with lysosome -> digestion
- In nutritional deprivation, autophagy is a survival mechanism
- If stress is not relieved, autophagy can trigger cell death
- Triggered by: nutrient deprivation, hypoxia, ER stress
4. Ferroptosis
- Cell death that depends on cellular iron levels
- Involves iron-dependent accumulation of lipid peroxides
- Distinct from apoptosis (not caspase-dependent), necrosis, and autophagy
- Regulated by GPX4 (glutathione peroxidase 4)
- Role in normal physiology and disease still under investigation
5. NETosis (Neutrophil Extracellular Trap-associated Cell Death)
- Unique to neutrophils
- On recognition of phagocytosed pathogens:
- Nuclear histone structures are modified
- Chromatin decondenses and nuclear membrane disrupts
- Decondensed chromatin enters cytoplasm and is released extracellularly
- Forms Neutrophil Extracellular Traps (NETs) - web-like structures
- NETs capture and kill bacteria, viruses, fungi, and parasites
- This form ("suicidal NETosis") results in neutrophil death
6. Entosis
- One cell actively internalizes another similar cell (a "cell within a cell")
- The swallowed cell remains alive in a vacuole and is eventually lysosomally degraded or released
- Regulated by cadherin-mediated cell-to-cell junctions
- Seen within epithelial cell populations; distinct from cell cannibalism in tumors
7. Paraptosis
- Induced by growth factor receptors (especially IGF-1 receptor)
- NOT caspase-dependent - mediated instead by MAP kinases
- Characterized by formation of large cytoplasmic vacuoles and mitochondrial swellings
- Morphologically distinct from apoptosis
8. Mitoptosis ("Mitochondrial Suicide")
- Programmed fragmentation and fusion of mitochondria with diminished ATP production
- Mitochondrial fragments are degraded by autophagic vacuoles or expelled as mitoptotic bodies
- Extensive mitochondrial fragmentation -> ATP failure -> cell death
Summary Table: Methods of Cell Death
| Type | Caspase? | Inflammation? | Membrane intact? | Key mediator |
|---|---|---|---|---|
| Apoptosis | Yes (3,7,8,9) | No | Yes | BCL-2/BAX, Fas/FasL |
| Necrosis | No | Yes | No (ruptured) | Ischemia, toxins |
| Necroptosis | No | Yes | No (ruptured) | RIP1/RIP3 kinases |
| Pyroptosis | Caspase-1 only | Yes (intense) | No | Inflammasome/IL-1β |
| Autophagy | No | Minimal | Yes | Autophagosome/lysosome |
| Ferroptosis | No | Variable | No | Iron/lipid peroxidation |
| NETosis | No | Yes | No | Neutrophil, chromatin NETs |
| Entosis | No | No | Variable | Cadherins |
| Paraptosis | No | No | Yes initially | MAPK, IGF-1R |
Sources:
- Robbins & Kumar Basic Pathology (Robbins Pathology) - primary reference for 2nd year MBBS
- Histology: A Text and Atlas with Correlated Cell and Molecular Biology
- Sleisenger & Fordtran's Gastrointestinal and Liver Disease
Exam tip: For a theory exam, always define apoptosis clearly, compare it with necrosis in a table, then describe both pathways in detail with their key molecules (BCL-2 family for intrinsic; Fas/FasL/FADD/caspase-8 for extrinsic). For "other methods," cover at least necroptosis, pyroptosis, autophagy, and ferroptosis - Robbins specifically mentions these four. NETosis, entosis, paraptosis, and mitoptosis are bonus marks.
Necrosis – a. Definition of Necrosis b. Classification, Etiopathogenesis & Morphology of Types of Necrosis
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
Loading Image
I now have all the textbook content, key concepts, and images. Here is the complete, exam-ready answer:
NECROSIS - Complete Answer for 2nd Year MBBS Theory Exam
(a) Definition of Necrosis
Necrosis is a form of pathological, irreversible cell death resulting from severe injury to the cell, characterized by:
- Denaturation of cellular proteins
- Enzymatic digestion of the lethally injured cell
- Disruption of the plasma membrane with leakage of cellular contents into the extracellular space
- Induction of inflammation in the surrounding tissue (this is the key distinguishing feature from apoptosis)
Necrosis is always pathological - it is caused by exogenous insults and is not a physiologically programmed process.
Causes of Necrosis:
- Ischemia (most common - loss of blood/oxygen supply)
- Physical agents: heat, cold, radiation, trauma
- Chemical agents: acids, alkalis, heavy metals, drugs
- Microbial toxins (bacterial, fungal)
- Immunologic injury
- Nutritional deficiency (extreme)
- In pancreatitis: active proteases leaking from damaged acinar cells
Pathogenesis of Necrosis (Etiopathogenesis)
The two fundamental and irreversible changes that define the "point of no return":
- Inability to reverse mitochondrial dysfunction - failure of oxidative phosphorylation and ATP generation even after the original injury resolves
- Profound disturbance of membrane function - injury to lysosomal membranes causes enzymatic dissolution of the cell
Sequence of events:
- Severe injury to cell
- Mitochondrial dysfunction -> ATP depletion
- Failure of ATP-dependent Na+/K+ pump -> Na+ and water influx -> cell swelling
- Anaerobic glycolysis -> lactic acid accumulation -> decreased intracellular pH
- Ribosomes detach from rough ER -> reduced protein synthesis
- Irreversible mitochondrial damage
- Lysosomal membrane rupture -> lysosomal enzymes enter cytoplasm -> autodigestion
- Plasma membrane disruption -> leakage of DAMPs (damage-associated molecular patterns: ATP, uric acid, HMGB1)
- DAMPs recognized by macrophage receptors -> recruitment of inflammatory cells
- Inflammatory cells release more proteolytic enzymes -> clearance of debris
Clinical significance of protein leakage:
- Cardiac muscle necrosis -> elevated cardiac Troponin in blood (detectable within 2 hours of MI)
- Hepatocyte necrosis -> elevated serum transaminases (AST, ALT)
- Bile duct necrosis -> elevated alkaline phosphatase
General Microscopic (Histological) Features of Necrosis
Cytoplasmic changes (H&E):
- Increased eosinophilia (pink staining) due to:
- Binding of eosin to denatured proteins
- Loss of basophilic RNA from cytoplasm
- Glassy, homogeneous appearance due to loss of glycogen
- Vacuolated, moth-eaten appearance when organelles are digested
- Myelin figures - whorled phospholipid precipitates replacing dead cells
- Discontinuities in plasma and organelle membranes (on EM)
Nuclear changes - THREE patterns (all due to DNA breakdown):
| Nuclear Change | Description |
|---|---|
| Pyknosis | Nuclear shrinkage + increased basophilia; DNA condenses into a dark, shrunken mass |
| Karyorrhexis | Fragmentation of the pyknotic nucleus |
| Karyolysis | Fading/dissolution of nuclear basophilia due to DNase digestion of DNA; complete disappearance within 1-2 days |
Memory tip: P-K-L = Shrinks, Fragments, Dissolves
(b) Classification, Etiopathogenesis & Morphology of Types of Necrosis
Robbins classifies necrosis into 6 morphological types:
- Coagulative Necrosis
- Liquefactive Necrosis
- Gangrenous Necrosis
- Caseous Necrosis
- Fat Necrosis
- Fibrinoid Necrosis
TYPE 1: COAGULATIVE NECROSIS
Most common type of necrosis
Etiology:
- Ischemia / infarction of solid organs (except brain)
- Examples: myocardial infarction, renal infarct, splenic infarct
Pathogenesis:
- Ischemia causes cell death
- The injury denatures both structural proteins AND enzymes (including proteolytic enzymes)
- Because proteolytic enzymes are denatured, autodigestion is blocked
- Tissue architecture is preserved for days to weeks
Gross morphology:
- Firm texture
- Pale/yellow-white infarct with well-defined margins
- e.g., wedge-shaped kidney infarct
Microscopic morphology:
- Preserved tissue architecture ("ghost outlines" of cells)
- Cells are eosinophilic and anucleate (loss of nucleus but cell shape intact)
- Inflammatory infiltrate at the margins
- Eventually, leukocytic enzymes digest the dead cells -> phagocytosis of debris
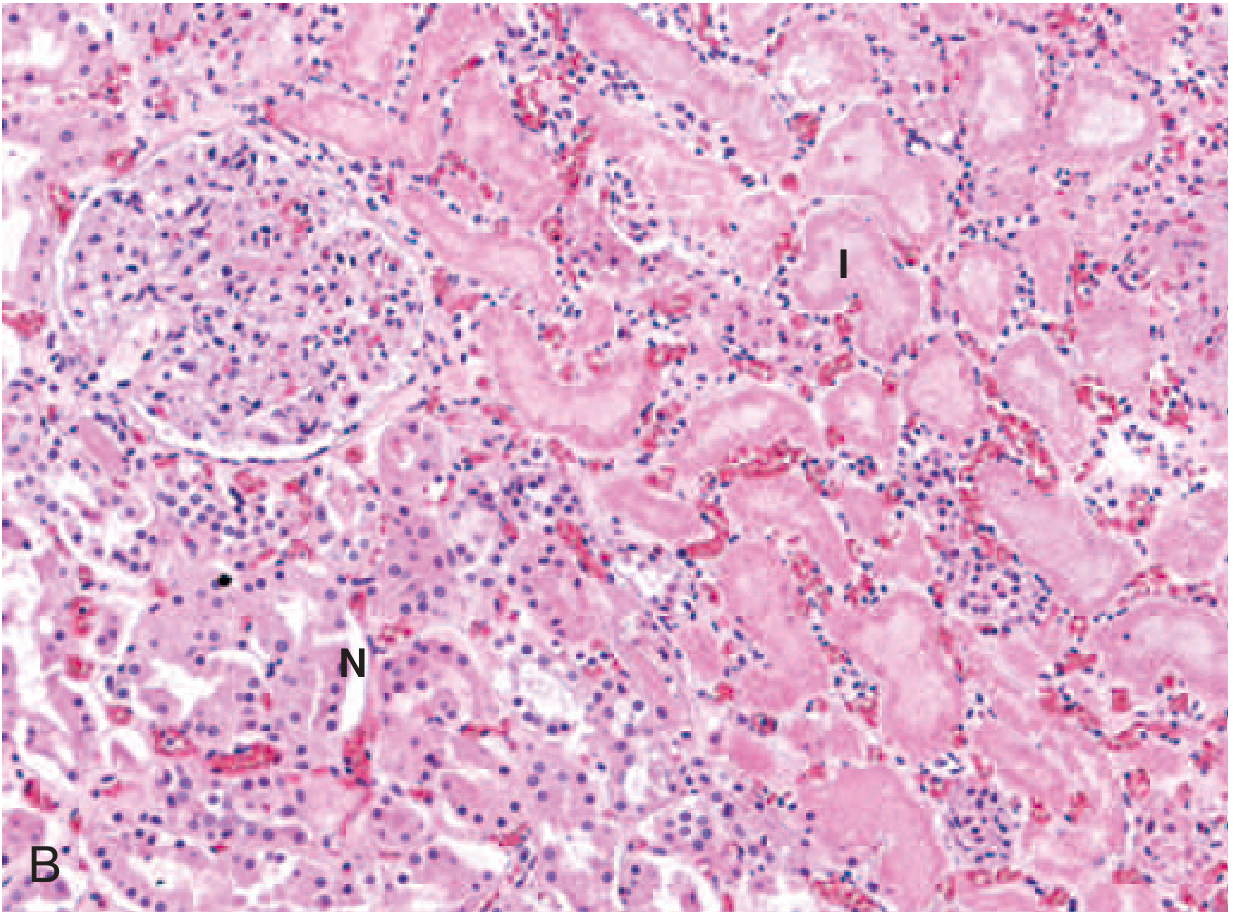
TYPE 2: LIQUEFACTIVE NECROSIS
Etiology:
- Bacterial or fungal infections (most common cause)
- Hypoxic death in the CNS/brain (most important exception to coagulative necrosis rule)
Pathogenesis:
- Microbes stimulate accumulation of neutrophils and macrophages
- Leukocytic enzymes (proteases, DNases, lipases) completely digest all cellular components
- In the brain: neurons and glial cells have large amounts of lipid and little structural protein; also rich in phospholipases -> rapid liquefaction
Gross morphology:
- Tissue is converted to a viscous liquid
- If due to bacterial infection: creamy yellow material = pus (due to leukocytes)
- A localized collection of pus = abscess
- In brain: soft, cystic cavity with yellow-brown fluid
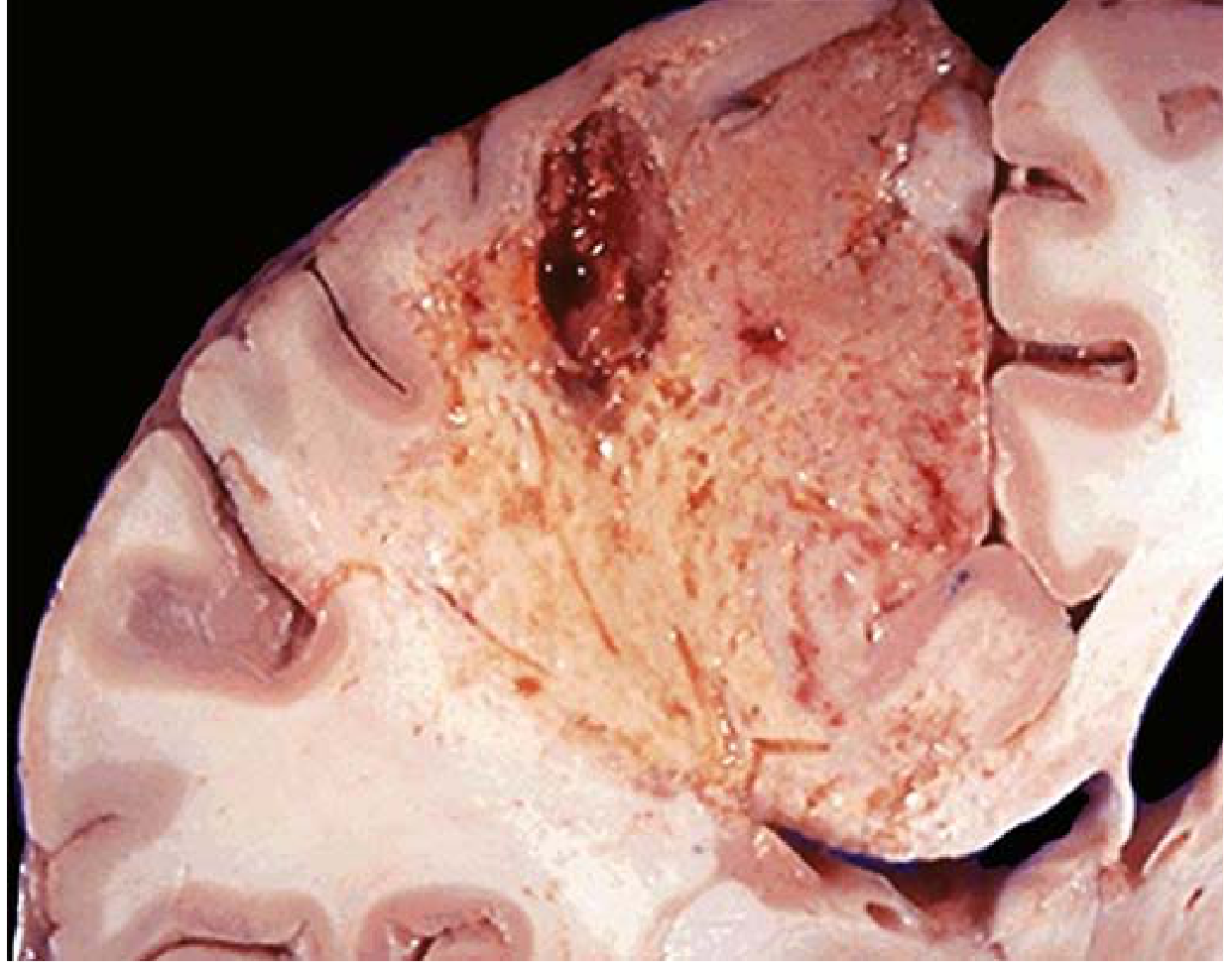
Microscopic morphology:
- Dead cells completely digested
- Abundant neutrophilic infiltrate
- Amorphous, granular, eosinophilic debris
- No preservation of tissue architecture
TYPE 3: GANGRENOUS NECROSIS
Note: Gangrenous necrosis is not a distinct microscopic pattern - it is a clinical term applied to a specific clinical situation
Definition: Refers to necrosis (usually coagulative) of a limb (most often lower leg) that has lost its blood supply, involving multiple tissue layers.
Types:
| Type | Features | Pathogenesis |
|---|---|---|
| Dry Gangrene | Mummified, dry, black, shrunken limb | Pure ischemic coagulative necrosis; no infection |
| Wet Gangrene | Moist, swollen, foul-smelling; rapid spread | Coagulative necrosis + superimposed bacterial infection -> secondary liquefactive changes |
| Gas Gangrene | Gas bubbles in tissue; crepitus on palpation; very rapid spread | Clostridium perfringens infection; releases gas (CO2, H2) via fermentation |
TYPE 4: CASEOUS NECROSIS
Etiology:
- Almost exclusively = Tuberculosis (Mycobacterium tuberculosis)
- Also: deep fungal infections (Histoplasma, Cryptococcus)
Pathogenesis:
- Granulomatous inflammation with central necrosis
- Activated macrophages (epithelioid cells) and T lymphocytes surround the necrotic center
- Cell-mediated delayed hypersensitivity (Type IV) plays a key role
- The lipid-rich mycobacterial cell wall and released toxins contribute to the distinctive appearance
Gross morphology:
- "Cheese-like" (caseous = Latin: cheese) - friable, yellow-white material
- Soft, crumbly consistency
- Classic appearance in pulmonary TB (Ghon focus/complex)
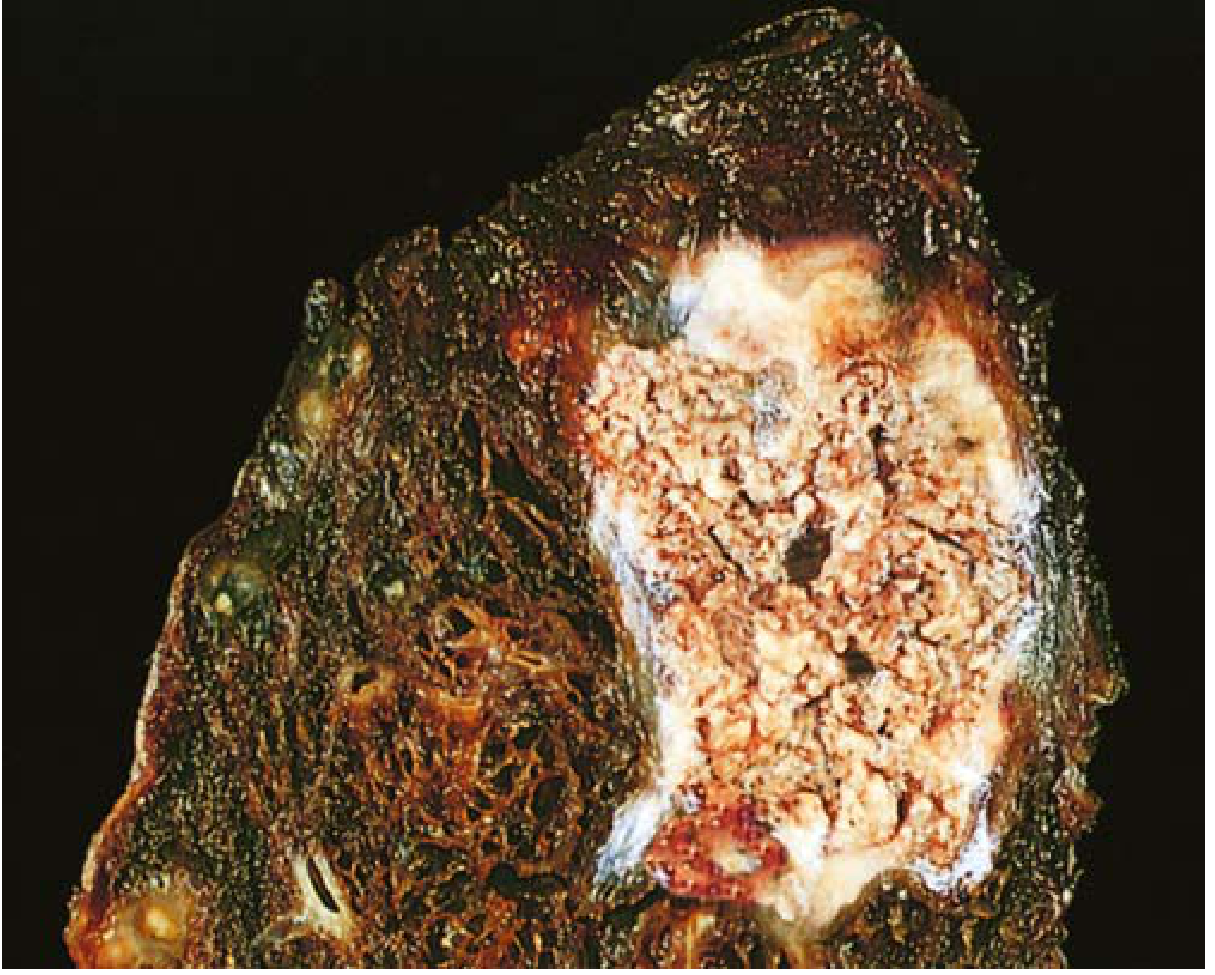
Microscopic morphology:
- Amorphous, granular, eosinophilic debris - no recognizable cell outlines
- Tissue architecture completely obliterated (unlike coagulative necrosis)
- Surrounded by a granuloma: epithelioid macrophages + Langhans giant cells + lymphocytes
- No neutrophils (unlike liquefactive)
Key distinction: Caseous necrosis = complete loss of architecture + surrounded by granuloma
TYPE 5: FAT NECROSIS
Etiology:
- Acute pancreatitis (most important cause)
- Abdominal/breast trauma
- Enzymatic: release of activated lipases from damaged pancreatic acinar cells
Pathogenesis:
- Activated pancreatic lipases (phospholipase A, lipase) leak from damaged pancreatic ducts and acinar cells
- Lipases digest peritoneal and mesenteric fat cells, releasing stored triglycerides
- Free fatty acids combine with calcium ions -> calcium soaps (saponification)
- This forms the chalky white deposits
Saponification = fatty acids + Ca²⁺ -> calcium soaps (chalky white lesions)
Gross morphology:
- Chalky white deposits on peritoneal/mesenteric fat
- "Candle-wax droplets" appearance
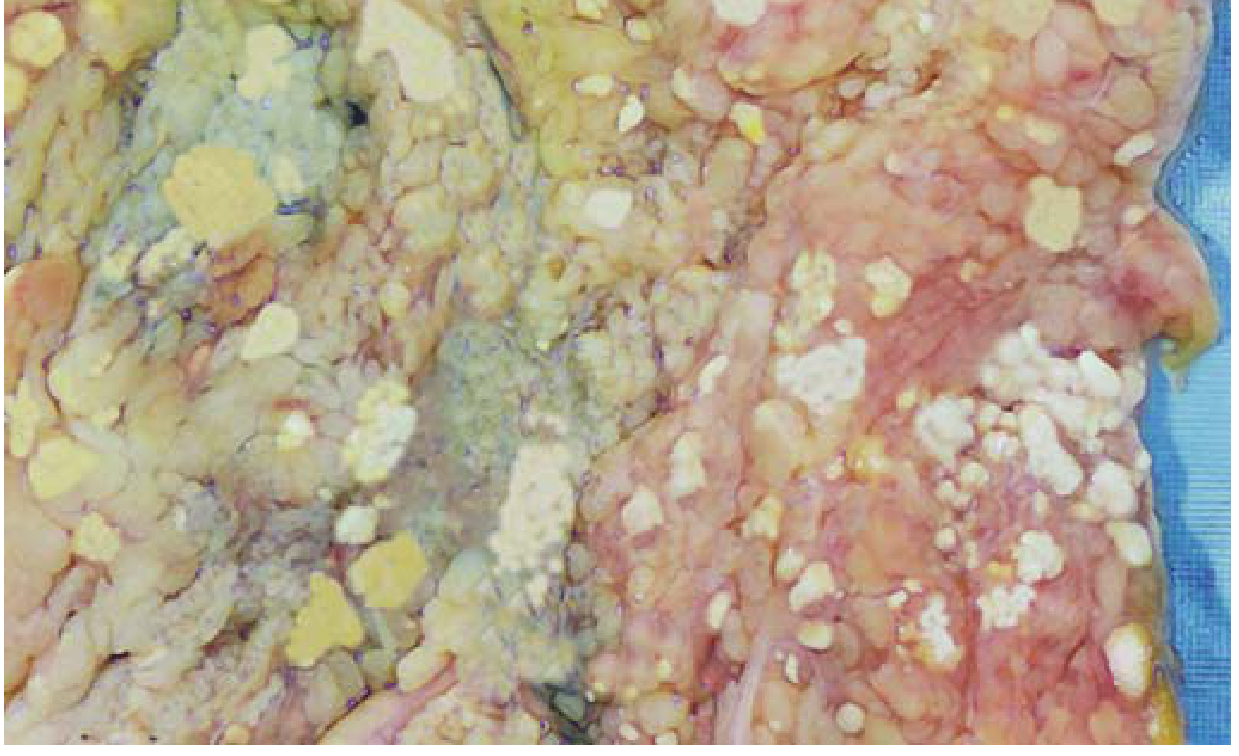
Microscopic morphology:
- Shadowy outlines of necrotic fat cells (ghost outlines of adipocytes)
- Surrounded by granular basophilic calcium deposits (bluish on H&E)
- Inflammatory reaction (macrophages, neutrophils) around the necrotic focus
TYPE 6: FIBRINOID NECROSIS
Etiology:
- Immune complex deposition in vessel walls (Type III hypersensitivity)
- Severe/malignant hypertension
- Autoimmune vasculitis (e.g., polyarteritis nodosa, SLE)
Pathogenesis:
- Antigen-antibody complexes deposit in the walls of small blood vessels
- Complement activation and neutrophil recruitment -> vessel wall injury
- Plasma proteins (especially fibrin) leak into vessel wall
- These proteins, plus necrotic vessel wall material, produce the characteristic appearance
Note: Fibrinoid necrosis is detected only by microscopy - no gross appearance
Gross morphology:
- Not visible grossly
Microscopic morphology:
- Bright pink (deeply eosinophilic), amorphous, homogeneous material in vessel walls on H&E
- The appearance mimics fibrin ("fibrin-like" = fibrinoid)
- Inflammatory infiltrate in and around vessel wall
- Loss of normal vessel wall architecture
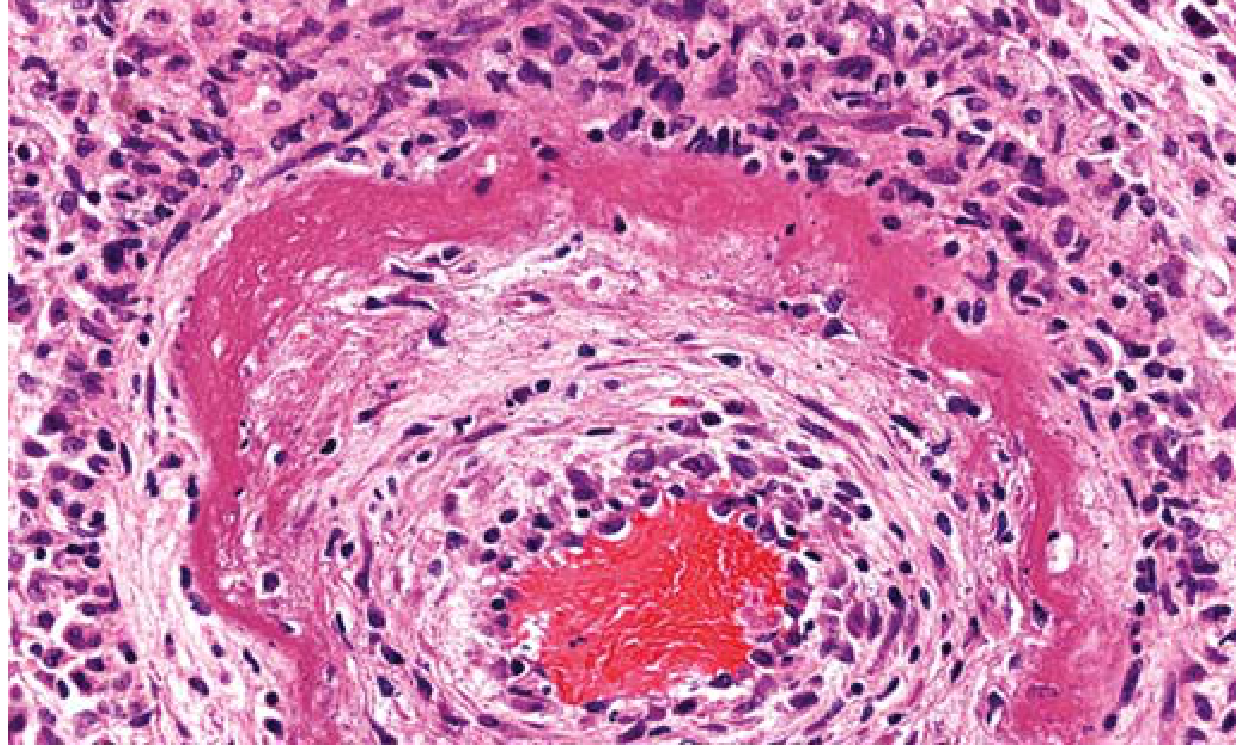
Summary Table: Types of Necrosis
| Type | Main Cause | Site | Gross | Microscopy | Key Feature |
|---|---|---|---|---|---|
| Coagulative | Ischemia/infarction | All solid organs except brain | Firm, pale, wedge-shaped | Ghost outlines, eosinophilic, anucleate cells | Architecture preserved |
| Liquefactive | Bacterial infection; CNS hypoxia | Brain, abscess | Pus (yellow); cystic cavity | Complete digestion, no architecture | Tissue liquefies |
| Gangrenous | Ischemia ± infection | Limbs | Dry (black/mummified) or Wet (moist/smelly) | Coagulative ± liquefactive | Clinical term, not microscopic type |
| Caseous | TB, fungi | Lung, lymph nodes | Friable yellow-white "cheese" | Amorphous granular debris + granuloma | Architecture obliterated + granuloma |
| Fat | Acute pancreatitis | Peritoneum, mesentery, breast | Chalky white deposits | Ghost fat cells + basophilic Ca²⁺ deposits | Saponification |
| Fibrinoid | Immune complex vasculitis, hypertension | Blood vessel walls | Not visible grossly | Bright pink amorphous material in vessel wall | Only seen microscopically |
Fate of Necrotic Cells
- Resolution - enzymatic digestion and phagocytosis by leukocytes (ideal outcome)
- Dystrophic calcification - calcium salts deposit in necrotic debris (especially if not cleared promptly)
- Organization and fibrosis - replacement by fibrous scar tissue
- Liquefaction and cyst formation - especially in brain
- Gangrenous transformation - if infected
Exam tip: For theory exams, always define necrosis clearly and contrast it with apoptosis in a sentence. Then for each type of necrosis, cover: (1) etiology, (2) pathogenesis in 2-3 lines, (3) gross morphology, and (4) microscopic morphology. Draw a summary table at the end - examiners love it. The most high-yield types are coagulative (most common), liquefactive (CNS/pus), caseous (TB), fat (pancreatitis), and fibrinoid (vasculitis).
Sources: Robbins & Kumar Basic Pathology; Robbins, Cotran & Kumar Pathologic Basis of Disease
This is a shared conversation. Sign in to Orris to start your own chat.