Alopecia detail and treatment guidelines from standard textbook for PG dermatology
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have very comprehensive content from all three major dermatology textbooks. Let me now write the full PG-level dermatology answer on alopecia.
Alopecia: Comprehensive Review for PG Dermatology
Sources: Fitzpatrick's Dermatology (9780071837781) | Dermatology 5e - Bolognia (9780702082252) | Andrews' Diseases of the Skin (9780323547536)
I. DEFINITION AND CLASSIFICATION
Alopecia refers to any form of hair loss. The primary classification divides it into:
Non-Scarring (Non-Cicatricial) Alopecia
Hair follicles are preserved and regrowth is possible.
- Androgenetic Alopecia (AGA)
- Alopecia Areata (AA)
- Telogen Effluvium (TE)
- Anagen Effluvium
- Traction Alopecia
- Tinea Capitis
- Trichotillomania
Scarring (Cicatricial) Alopecia
The follicular bulge (stem cell reservoir) is destroyed by fibrosis - hair loss is permanent.
Per the North American Hair Research Society (NAHRS) classification:
| Type | Entities |
|---|---|
| Lymphocytic | Lichen Planopilaris (LPP), Frontal Fibrosing Alopecia (FFA), Graham Little syndrome, Fibrosing Alopecia in Pattern Distribution (FAPD), Classic Pseudopelade (pseudopelade of Brocq), Central Centrifugal Cicatricial Alopecia (CCCA), DLE, Alopecia Mucinosa |
| Neutrophilic | Folliculitis Decalvans, Dissecting Cellulitis (perifolliculitis capitis abscidens et suffodiens) |
| Mixed | Acne Keloidalis, Folliculitis necrotica, Erosive Pustular Dermatosis |
| Secondary | Burns, radiation, malignancy, sarcoidosis, morphea, cutaneous TB |
- Dermatology 5e (Bolognia), p. 1404
II. HAIR CYCLE - PHYSIOLOGICAL BASIS
Understanding hair cycling is fundamental to all alopecias:
- Anagen (growth): ~90% of scalp hairs; lasts 2-4 years
- Catagen (involution): 0.5-1% of follicles
- Telogen (resting/shedding): ~10% of follicles; lasts ~3 months
- Normal daily shedding: 50-200 hairs
- Total scalp hair follicles: ~100,000
III. ANDROGENETIC ALOPECIA (AGA)
Pathogenesis
AGA is a nonscarring, progressive miniaturization of hair follicles - conversion of terminal pigmented anagen hairs to fine hypopigmented vellus hairs. The key hormone is dihydrotestosterone (DHT), formed by the action of 5-alpha-reductase (3 isoenzymes: I, II, III) on testosterone. DHT mediates perifollicular fibrosis, hair sheath thickening, and progressive follicle miniaturization. The enzyme 5-alpha-reductase is increased in scalp of balding individuals.
Genetic basis:
- Polygenic inheritance (polygenic, not simple Mendelian)
- The androgen receptor gene (AR) on the long arm of X chromosome (Xq11-12) is the major determinant in men
- AR-CAG polymorphism: fewer CAG repeats = greater androgen sensitivity = higher AGA risk and better finasteride response
- Men with the G allele of rs6152 have 70% lifetime risk; A allele = low risk
- Other genes: EDA2R (X-linked), WNT10A (bulge expression), APCDD1 (WNT inhibitor), CYP19A1, ESR2 (implicated in female AGA)
- Eunuchs do not develop baldness unless given androgen therapy
- In congenital 5-alpha-reductase type II deficiency, baldness does not occur
Hair cycle changes: Progressive shortening of anagen phase, increasing telogen proportion, lengthening of kenogen (lag between telogen and anagen), and progressive shaft diameter reduction.
Histology:
-
Decrease in anagen/increase in telogen follicles
-
Follicular miniaturization and anisotrichosis (variability in shaft diameter) - best seen on transverse sections
-
Fibrous tract remnants ("streamers") below miniaturized follicles
-
Many mast cells in fibrous tracts; inflammatory cells absent
-
Stem cells preserved in bald scalp (progenitor cells are reduced - key finding)
-
Fitzpatrick's Dermatology, Ch. 85 | Andrews', p. 872 | Dermatology 5e (Bolognia), p. 3158
Clinical Patterns
Male Pattern Hair Loss - Hamilton-Norwood Classification
Frontal hairline recedes bilaterally at the Geheimratswinkeln ("professor angles"), followed by vertex thinning, progressively until the entire top is bald. The parietal and occipital areas are permanently spared (permanent donor zone). The Hamilton-Norwood classification (Grades I-VII) describes progressive stages.
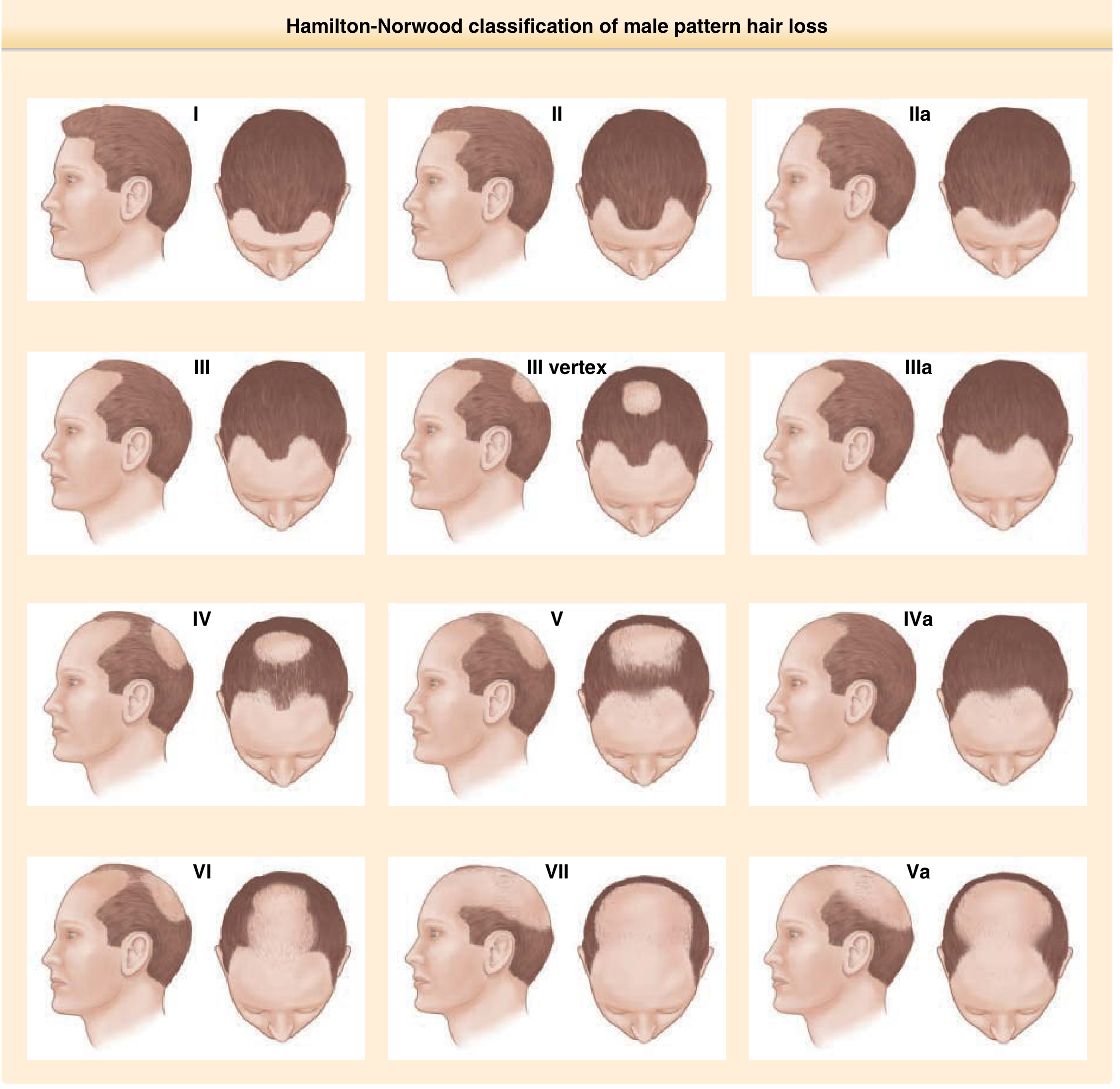
Female Pattern Hair Loss (FPHL) - Ludwig Classification
-
Diffuse centroparietal thinning with preservation of the frontal hairline (distinguishes it from male pattern)
-
Ludwig scale: 3-point; Sinclair scale: 5-point
-
Christmas tree pattern: centroparietal thinning with additional breaching of the frontal hairline
-
Role of androgens less certain in women; subset have associated hormonal dysregulation
-
Associated genes: CYP19A1 and ESR2 polymorphisms
-
Fitzpatrick's Dermatology, p. 1526
Diagnosis of AGA
- Clinical: history + pattern examination; hair pull test
- Hair pull test (Sabouraud maneuver): Grasp 50-60 hairs; pull firmly. >10% pulled = positive (confirms active shedding). In AGA, positive frontally, negative occipitally.
- Dermoscopy/Trichoscopy: hair diameter variations, loss of trio-hair groups with single terminal hairs, increased vellus hairs, absent follicular ostia (excludes scarring alopecia)
- Biopsy rarely indicated; use if differential diagnosis includes cicatricial alopecia or diffuse AA
- Lab workup in women: ferritin, TSH; if hyperandrogenism signs - full endocrine workup
Treatment of AGA
First-Line Medical Treatments
1. Topical Minoxidil
- Available as 2% and 5% solutions and 5% foam (both OTC)
- Promotes dermal papilla cell survival, prolongs anagen phase, enlarges shaft diameter
- Best results in: early cases (<10 years), limited extent (bald area <10 cm vertex), pretreatment density >20 hairs/cm²
- Must be used indefinitely to maintain response
- Note: initial telogen effluvium may occur (premature termination of telogen triggering new anagen)
- Low-dose oral minoxidil is also a recognized option (particularly for female AGA)
2. Oral Finasteride (1 mg daily) - for men
- Type 2, 5-alpha-reductase inhibitor
- Effective in men aged 18-41 with mild-to-moderate hair loss at vertex, anterior midscalp, and frontal region
- Stops hair loss in up to 90% of men for 5+ years; ~65% demonstrate hair regrowth
- Requires 6+ months for visible response; 12 months to assess failure
- Hair patterning on temples not improved
- Contraindicated in women who may become pregnant
- Long-term use required; regimens combining finasteride + minoxidil more effective than either alone
3. Dutasteride
- Blocks both type 1 and type 2 5-alpha-reductase (broader than finasteride)
- Effective in male-pattern hair loss; also used in FFA
Emerging and Adjunct Treatments
| Treatment | Evidence / Notes |
|---|---|
| Platelet-Rich Plasma (PRP) | Role being defined; preliminary studies promising |
| Microneedling | Often combined with minoxidil; role being established |
| Low-Level Laser Therapy (LLLT) | LaserComb/helmet; inconsistent results, low evidence |
| Prostaglandin D2 inhibitors | PGD2 inhibits hair growth (via DP2 receptor on balding scalp); therapeutic target |
| Topical adenosine | Some promise in preliminary studies |
| Spironolactone (women) | Anti-androgen effect for FPHL |
| Topical caffeine, melatonin, retinoids | Supportive; limited controlled data |
| Biotin, zinc, micronutrients | No robust evidence unless nutritional deficiency present |
Surgical Treatment
-
Hair Transplantation: Follicular units of 1-4 hairs transplanted. Two major methods:
- FUT (Follicular Unit Transplantation): elliptical donor strip harvesting
- FUE (Follicular Unit Excision): individual follicle extraction (less invasive)
-
Indicated for stable, non-progressive AGA with sufficient donor hair density
-
Combination with finasteride 1mg +/- topical minoxidil recommended to reduce postoperative AGA progression
-
First hair transplant procedure: 1950s (large punch autografts)
-
Andrews', p. 872 | Fitzpatrick's Dermatology, p. 1533-34
IV. ALOPECIA AREATA (AA)
Pathogenesis
AA is an autoimmune, non-scarring alopecia characterized by intermittent and/or chronic hair loss. Key mechanisms:
- Loss of immune privilege of the hair follicle: HF normally expresses low MHC Class I/II antigens and produces immunosuppressive cytokines/neuropeptides
- CD8+ NKG2D+ cytotoxic T cells are primary effectors; CD4+ helper T cells promote, Treg cells (CD4+CD25+) suppress disease
- Key cytokines: IFN-gamma, IL-2, IL-15 (CD8+ T cell induction/persistence)
- JAK/STAT pathway is critical - JAK1/2 inhibition is therapeutic
- GWAS-identified associations: CTLA4, ICOS, IL21/IL2, IL2RA, NKG2D ligands, HLA-DR/DQ (dominant susceptibility locus)
- IL-15 neutralization attenuates disease in animal models
Hair cycle impact: CD8+ T cells induce premature catagen via IFN-gamma, causing HF dystrophy. T cell inflammation preferentially targets anagen hair bulbs (not the bulge), explaining why AA is nonscarring and regrowth is possible.
- Fitzpatrick's Dermatology, p. 229-230
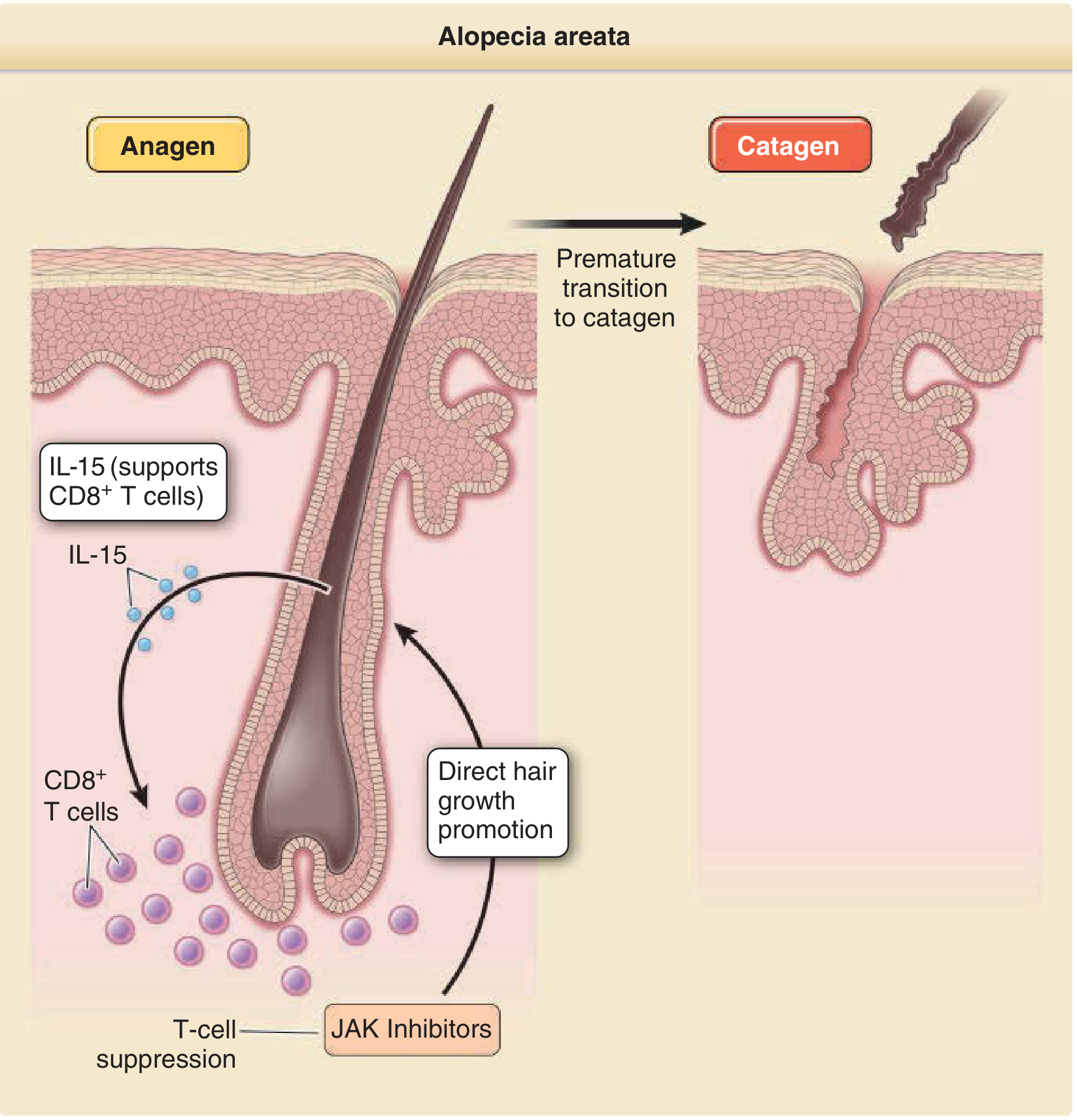
Clinical Features
- Patchy AA (AAP): well-defined, smooth oval/round patches of hair loss; exclamation-mark hairs at periphery
- Alopecia Totalis (AT): entire scalp hair loss
- Alopecia Universalis (AU): entire scalp + body hair loss
- Nail changes: fine pitting, trachyonychia (20-nail dystrophy)
- Associated: thyroiditis, other autoimmune diseases
- Positive hair pull test at active margins
Histology (Key for exams):
-
Early/active disease ("swarm of bees"): peribulbar mononuclear cell infiltrate (lymphocytes + occasional eosinophils) around terminal anagen/catagen bulbs; exocytosis into bulbar epithelium; trichomalacia; pigment casts
-
Chronic/stable disease: majority in catagen/telogen; diffuse miniaturization; numerous "nanogen" (rapidly cycling miniaturized) hairs; mild peribulbar infiltrate
-
Dermatology 5e (Bolognia), p. 3052-73
Treatment of Alopecia Areata
| Treatment | Route | Notes |
|---|---|---|
| Intralesional corticosteroids (triamcinolone acetonide 5-10 mg/mL) | Intralesional | First-line for patchy AA; injections every 4-6 weeks |
| Topical corticosteroids | Topical | Clobetasol; for limited disease, especially in children |
| Systemic corticosteroids | Oral/IV | For rapidly progressive/extensive disease; high relapse rate on discontinuation |
| Contact immunotherapy (DPCP, SADBE) | Topical | For extensive/refractory disease; induces Th2 response |
| JAK inhibitors (Baricitinib, Ritlecitinib) | Oral | Major advance; FDA approved (ritlecitinib for AA, 2023); target JAK1/2 signaling; reverse hair loss by suppressing inflammation and directly acting on hair follicles to induce growth |
| Minoxidil | Topical | Adjuvant to promote regrowth |
| Anthralin | Topical | Short-contact therapy |
| PUVA / Narrowband UVB | Phototherapy | Limited efficacy; relapse common |
| Methotrexate | Systemic | For severe/refractory disease |
| Cyclosporine | Systemic | Effective but high relapse rate |
V. TELOGEN EFFLUVIUM (TE)
Types and Pathophysiology
TE is increased shedding of otherwise normal telogen club hairs due to premature or excessive conversion of anagen follicles to telogen. It occurs 3-5 months after the inciting cause (reflecting the telogen phase duration).
Five pathophysiological mechanisms (Headington's classification):
- Immediate anagen release (premature, sudden termination of anagen)
- Delayed anagen release (post-pregnancy: prolonged anagen released simultaneously)
- Short anagen syndrome (shortened anagen phase)
- Immediate telogen release (prolonged telogen curtailed by systemic trigger)
- Delayed telogen release (synchronization of telogen leading to massive shedding)
Common triggers:
- Surgery, parturition, febrile illness, crash dieting, medications (heparin, retinoids, beta-blockers, carbamazepine, anticoagulants, hormonal contraceptives)
- Hypothyroidism, iron deficiency (low ferritin), zinc deficiency, protein malnutrition
- Lupus erythematosus, inflammatory scalp disorders, significant emotional stress
- SARS-CoV-2 infection (quarter of infected patients developed TE within 2 months)
- Note: Topical minoxidil initiation can cause transient TE (premature termination of telogen to initiate anagen)
Chronic Telogen Effluvium (CTE):
- Idiopathic diffuse club hair loss in middle-aged women (4th-6th decade)
- Telogen shedding >6 months; fluctuating course
- Patients typically have denser-than-average hair before onset; marked bitemporal recession
- Positive pull test at both vertex AND occiput (distinguishes from AGA, which is frontal only)
Histology:
- Normal total number of hairs
- Normal terminal (large) hair number
- Telogen count >20% (>15% is suggestive); rarely >50%; >80% inconsistent with TE
- No inflammation, no scarring
Diagnosis
- Hair pull test: >4-6 club hairs (with visible depigmented club-shaped bulb, no sheath)
- Trichogram (50 hairs plucked with Kelly clamp): anagen:telogen ratio
- Lab: CBC, ferritin, TFTs, zinc, ANA
- Normal 100-150 hairs shed daily; TE: 150-400+ hairs/day
Treatment
- Identify and treat the underlying cause
- Reassurance (most acute TE resolves within 6 months)
- Nutritional supplementation if deficient (iron, zinc, biotin)
- Topical minoxidil if prolonged/chronic
- Treat concurrent scalp conditions (seborrheic dermatitis)
VI. CICATRICIAL (SCARRING) ALOPECIAS - Key Entities
Lichen Planopilaris (LPP)
- Pathophysiology: Lymphocytic infiltrate targeting the infundibulo-isthmic (bulge) region - irreversible follicle destruction
- Demographics: Predominantly White women, 40-60 years
- Clinical: Progressive scalp pruritus/burning, perifollicular erythema and perifollicular scaling, small patches coalescing to larger areas of cicatricial alopecia; NO hair follicle openings
- Trichoscopy: Perifollicular scaling, absence of follicular openings
- Histology: Lymphocytic infiltrate at isthmus and infundibulum; fibrosis
- Treatment: Hydroxychloroquine (first-line systemic), intralesional/topical corticosteroids, cyclosporine, mycophenolate mofetil, pioglitazone (PPARgamma agonist)
Frontal Fibrosing Alopecia (FFA)
- Band-like frontotemporal recession with loss of eyebrows, perifollicular erythema and hyperkeratosis
- May have "lonely hairs" at the hairline margin, glabellar red follicular dots, facial papules
- May coexist with classic lichen planus or lichen planus pigmentosus
- Treatment: Similar to LPP; oral finasteride or dutasteride may be beneficial (reduces vellus hair follicle proportion, the primary target in FFA)
Graham Little-Piccardi-Lasseur Syndrome
Triad: (1) Lichen planopilaris of scalp + (2) non-cicatricial alopecia of axilla and groin + (3) follicular lichen planus of the body
Central Centrifugal Cicatricial Alopecia (CCCA)
- Demographics: Most commonly Black women; some patients have PADI3 mutations
- Slowly progressive, symmetric cicatricial alopecia centered on mid-scalp/vertex
- Treatment: Topical/intralesional corticosteroids, oral tetracyclines, adjuvant immunosuppressives
Folliculitis Decalvans (Neutrophilic)
- Recurrent pustules around follicles, progressive scarring; Staphylococcus aureus implicated
- Treatment: prolonged rifampicin + clindamycin combination; doxycycline
Dissecting Cellulitis of the Scalp (Neutrophilic)
- Deep follicular and perifollicular abscesses, sinuses, scarring alopecia; mainly Black men
- Part of follicular occlusion tetrad
- Treatment: isotretinoin; biologics (adalimumab) for refractory disease
VII. TRACTION ALOPECIA
- Caused by chronic mechanical tension on the hair shaft (tight braiding, extensions, tight hairstyles)
- Initially reversible, becomes permanent if tension continues
- Marginal pattern (fronto-temporal) in braiding-related traction
- Treatment: Cease offending hairstyle; topical minoxidil in early stages
VIII. CLINICAL APPROACH TO ALOPECIA
Key Historical Points
- Shedding (telogen effluvium) vs. reduced density only (miniaturization/AGA) vs. breakage
- Rate of onset, duration, pattern distribution
- Scalp symptoms: pruritus, burning, pain (active cicatricial)
- Precipitating events (surgery, fever, delivery, stress, medication changes)
- Family history; medications; nutritional status
- Signs of hyperandrogenism in women (acne, hirsutism, irregular menses)
Diagnostic Algorithm
Alopecia
├── Scarring (no follicular openings, fibrosis)
│ ├── Lymphocytic → LPP, FFA, DLE, CCCA
│ ├── Neutrophilic → Folliculitis decalvans, Dissecting cellulitis
│ └── Mixed → Acne keloidalis
└── Non-Scarring
├── Diffuse
│ ├── With precipitant → Telogen Effluvium
│ ├── Patterned → AGA
│ └── Diffuse, no precipitant → Chronic TE / Diffuse AA
└── Patchy
├── Smooth patches + exclamation hairs → Alopecia Areata
├── Broken hairs of varying length → Trichotillomania
└── Scaling + inflammation → Tinea Capitis
Biopsy Guidelines
- 4mm punch biopsy at hair-bearing margin (guided by trichoscopy), extending into subcutaneous fat
- Two biopsies recommended: one for vertical sectioning, one for horizontal sectioning
- Do NOT biopsy the central scarred zone (only end-stage fibrosis will be seen)
IX. SUMMARY TABLE: KEY DIFFERENTIALS
| Feature | AGA | AA | Telogen Effluvium | LPP/FFA |
|---|---|---|---|---|
| Scarring | No | No | No | Yes |
| Pattern | Vertex/Frontal (M); Centroparietal (F) | Patchy/Diffuse | Diffuse | Frontotemporal (FFA); Vertex (LPP) |
| Scalp symptoms | None | None | None | Burning, pruritus, pain |
| Exclamation hairs | No | Yes | No | No |
| Pull test | + frontal (AGA) | + active margin | + diffuse | Variable |
| Key histology | Miniaturization, fibrous streamers | Peribulbar lymphocytes ("swarm of bees") | Increased telogen >20% | Lymphocytic infiltrate at bulge |
| Primary treatment | Minoxidil, Finasteride | Intralesional steroids, JAK inhibitors | Treat cause | Hydroxychloroquine, steroids |
Sources: Fitzpatrick's Dermatology, Ch. 13, 85, 86 | Andrews' Diseases of the Skin, Ch. 33 | Dermatology 5e (Bolognia), Ch. 69, 157
Give me list of all lesions of dermatology with pic from derma net and describe the lesions as well. It should serve as guide for my derma posting
Asking for Preferences
Reading Web Page
dermatology lesion description clinical examples images
Reading Web Page
dermatology lesion description clinical examples
Searching the Web
dermnetnz.org macule papule plaque vesicle dermatology lesions image gallery site:dermnetnz.org
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Searching the Web
site:dermnetnz.org/assets lesion macule vesicle bulla pustule wheal images jpg
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Your complete PG Dermatology Posting Guide is ready. Here's a summary of what's covered:
What's in this guide (30 sections):
Part A - Primary Lesions (12 lesions):
Macule, Patch, Papule, Plaque, Nodule, Tumour, Vesicle, Bulla, Pustule, Wheal, Cyst, Comedone - each with definition, size, palpability, contents, key teaching points, and clinical disease examples.
Part B - Secondary Lesions (9 lesions):
Scale (with 7 scale types + diseases), Crust, Erosion, Ulcer (with 5 border types + leg ulcer classification), Excoriation, Fissure, Lichenification, Scar, Atrophy.
Part C - Special/Vascular Lesions (5):
Telangiectasia, Purpura/Petechiae/Ecchymosis, Milia, Abscess, Burrow.
Part D - Diagnostic Extras:
- Lesion configurations (annular, targetoid, herpetiform, Koebner, sporotrichoid, Blaschkolinear, etc.)
- Surface topography terms (lichenoid, umbilicated, verrucous, pedunculated)
- Color significance guide
- 10 key clinical signs (Nikolsky, Auspitz, Darier's, Asboe-Hansen, etc.)
- Quick-reference summary table of all lesions
Note: DermNet's website is blocking direct image extraction, so images are linked directly to DermNet pages with illustrative URLs - clicking any image will take you to the full DermNet article for that condition.
Give me pic of all dermatology lesions and diagnosis from standard textbooks
Finding Sources
Reading File
Searching Content
Searching Content
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Loading Image
Loading Image
Loading Image
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
Loading Image
Loading Image
Loading Image
Loading Image
Finding Sources
Reading File
Loading Image
Loading Image
Here is your complete visual dermatology lesion gallery with real textbook photographs and clinical diagnoses, sourced from Andrews' Clinical Dermatology Atlas 2e and Dermatology 5e (Boliviana).
Visual Dermatology Lesion Gallery
With Textbook Clinical Photographs
Source: Andrews' Clinical Atlas of Skin Disease (9780323790130) | Dermatology 5e - Boliviana (9780702082252)
PRIMARY LESIONS
1. MACULE
Definition: Flat, non-palpable, circumscribed color change < 1 cm
Nevus of Ota (macule)
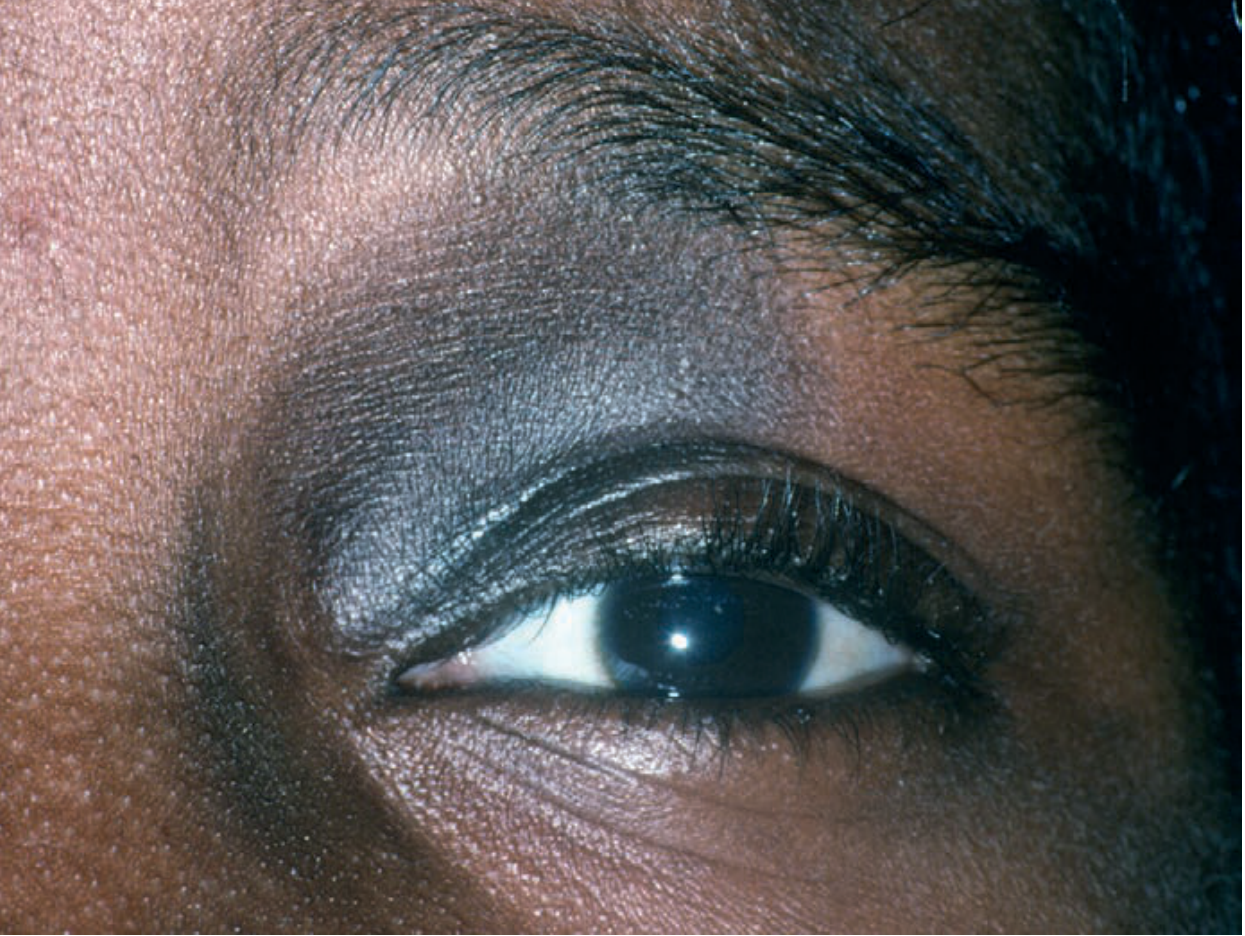 Fig. 2.1 - Blue-gray macule periorbital distribution due to dermal melanocytosis. Courtesy Steven Binnick, MD. Source: Andrews' Clinical Atlas.
Fig. 2.1 - Blue-gray macule periorbital distribution due to dermal melanocytosis. Courtesy Steven Binnick, MD. Source: Andrews' Clinical Atlas.
Diagnosis clue: Blue-gray color = dermal melanin location. Unilateral periorbital + scleral involvement = Nevus of Ota (oculodermal melanocytosis). Risk of glaucoma and melanoma.
Voriconazole-induced lentigines (macules)
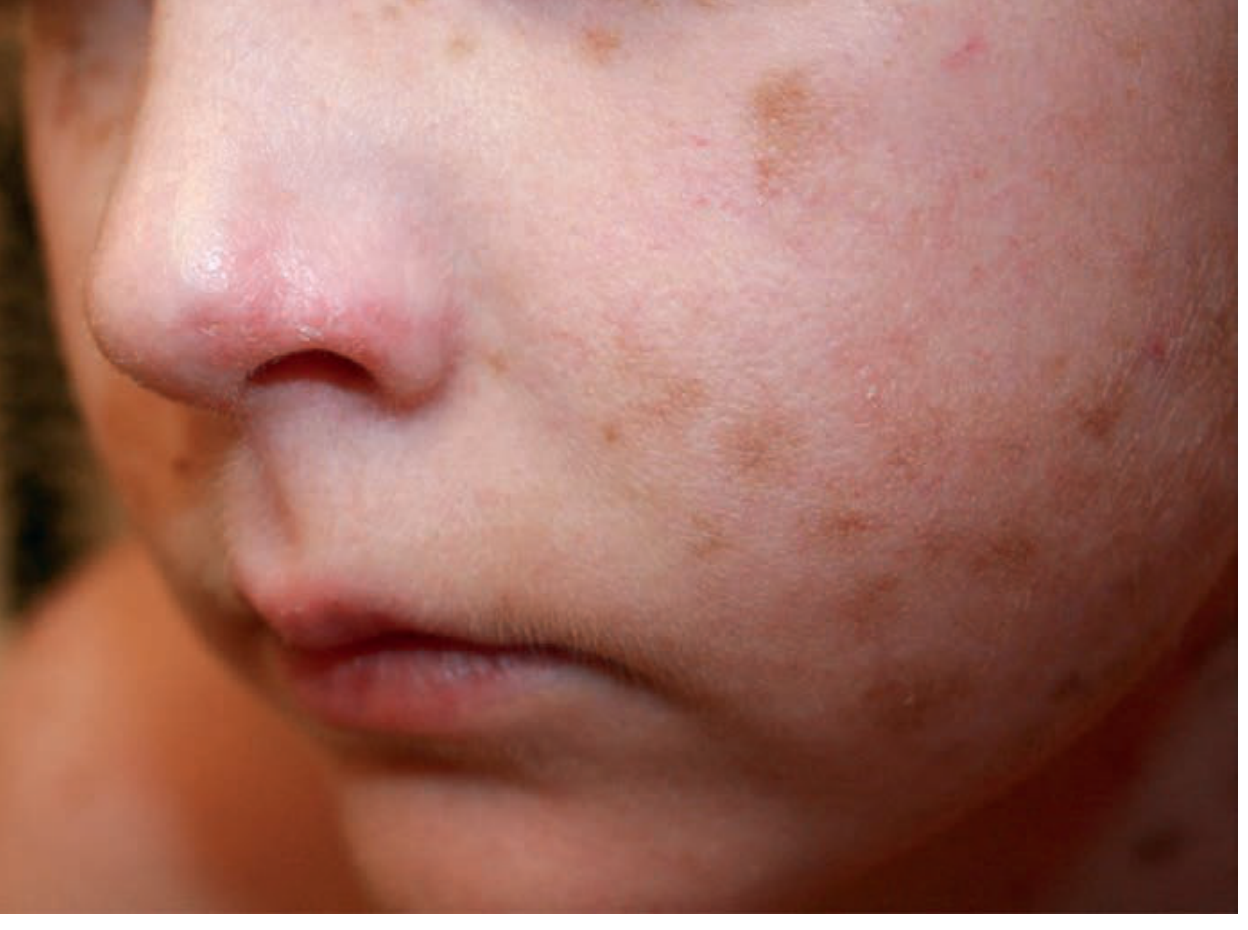 Fig. 2.3 - Multiple small hyperpigmented macules (lentigines) on the face, drug-induced. Courtesy Jennifer Huang, MD. Source: Andrews' Clinical Atlas.
Fig. 2.3 - Multiple small hyperpigmented macules (lentigines) on the face, drug-induced. Courtesy Jennifer Huang, MD. Source: Andrews' Clinical Atlas.
Diagnosis clue: Multiple discrete brown macules on sun-exposed face. Voriconazole (antifungal) causes photocarcinogenesis and lentigines - ask about azole use in transplant patients.
Axillary freckling in Neurofibromatosis Type 1 (macules)
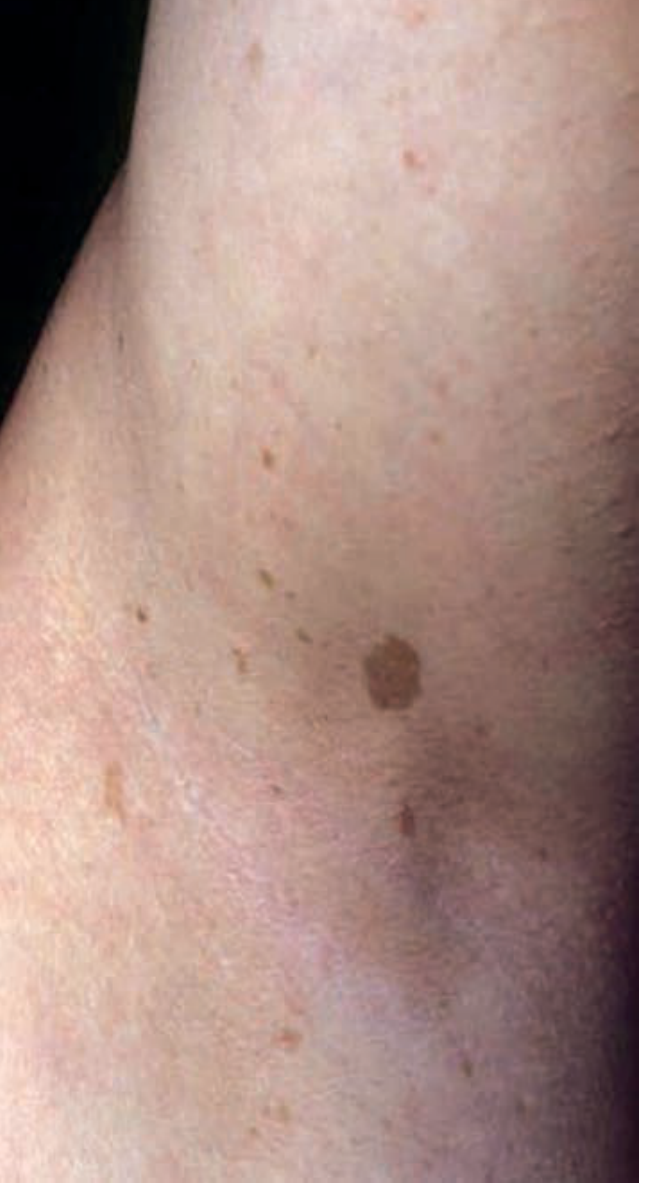 Fig. 2.2 - Multiple discrete hyperpigmented macules in axilla (Crowe's sign). Source: Andrews' Clinical Atlas.
Fig. 2.2 - Multiple discrete hyperpigmented macules in axilla (Crowe's sign). Source: Andrews' Clinical Atlas.
Diagnosis clue: Axillary/inguinal freckling (Crowe's sign) = pathognomonic for NF1. Along with ≥6 café-au-lait macules (>5mm prepubertal, >15mm postpubertal) and Lisch nodules (iris hamartomas). NF1 diagnostic criteria require 2/7 features.
2. PATCH
Definition: Flat, non-palpable, circumscribed color change > 1 cm
Vitiligo (patch)
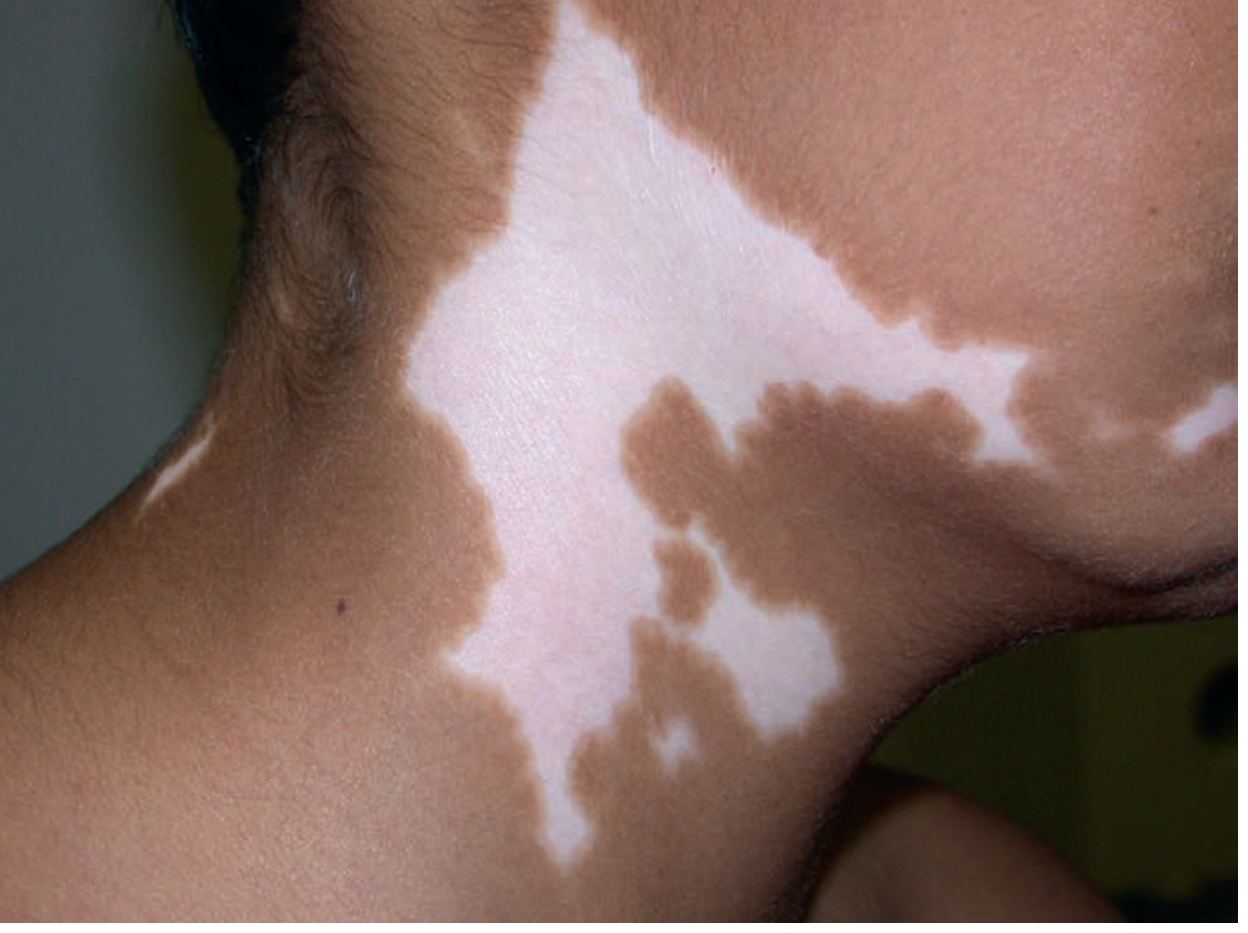 Fig. 2.4 - Large depigmented white patches on the neck. Source: Andrews' Clinical Atlas.
Fig. 2.4 - Large depigmented white patches on the neck. Source: Andrews' Clinical Atlas.
Diagnosis clue: Chalk-white depigmented (not hypopigmented) patches with well-defined borders. Autoimmune destruction of melanocytes. Wood's lamp: chalk-white fluorescence. Associated with thyroiditis, DM, Addison's. Treatment: tacrolimus, NB-UVB, JAK inhibitors (ruxolitinib cream FDA approved).
Nevus Depigmentosus (patch)
 Fig. 2.5 - Hypopigmented (not depigmented) patch, irregular borders, stable from birth. Source: Andrews' Clinical Atlas.
Fig. 2.5 - Hypopigmented (not depigmented) patch, irregular borders, stable from birth. Source: Andrews' Clinical Atlas.
Diagnosis clue: Hypopigmented (NOT white) patch, present since birth, stable, irregular "moth-eaten" borders. Melanocytes present but functionally reduced. Wood's lamp: does NOT fluoresce chalk-white (unlike vitiligo). Does NOT repigment.
3. PAPULE
Definition: Elevated, palpable, solid, circumscribed < 1 cm
Eruptive Xanthomas (papules)
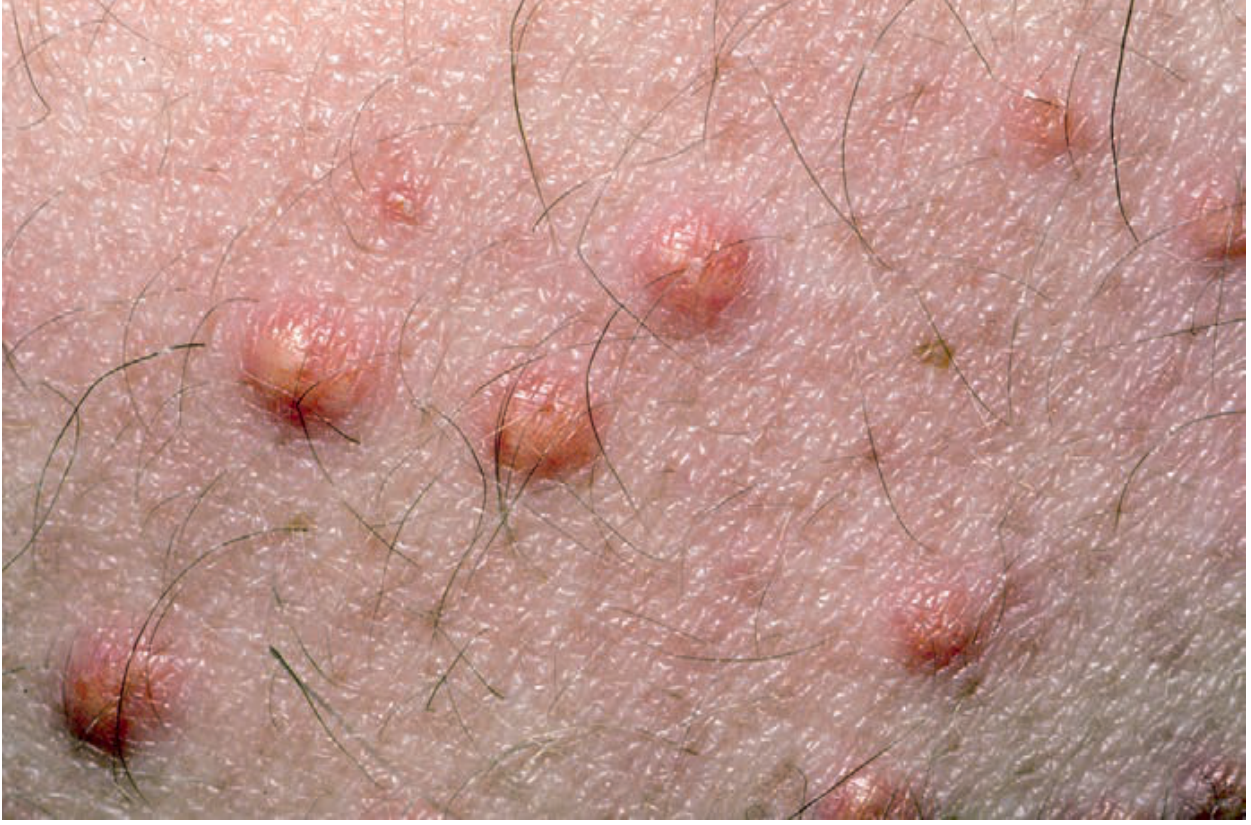 Fig. 2.9 - Multiple yellowish papules. Yellow color helps narrow the differential. Source: Andrews' Clinical Atlas.
Fig. 2.9 - Multiple yellowish papules. Yellow color helps narrow the differential. Source: Andrews' Clinical Atlas.
Diagnosis clue: Yellow papules with erythematous halo on extensor surfaces, buttocks. Sudden-onset = very high triglycerides (>1000 mg/dL). Associated with: diabetes, hypothyroidism, pancreatitis risk. Treat: triglyceride reduction (fibrates, dietary). Yellow skin color: xanthoma, jaundice, carotenemia.
4. PLAQUE
Definition: Elevated, palpable, flat-topped, > 1 cm; formed by papule coalescence
Classic plaque examples on the ward:
- Psoriasis: Silver-white micaceous scale on erythematous plaques; Auspitz sign; extensor surfaces, scalp, nails
- Lichen simplex chronicus: Lichenified plaques from chronic rubbing; posterior neck, legs
- Mycosis fungoides: Indurated erythematous plaques (cutaneous T-cell lymphoma); "patch → plaque → tumour" progression
- Morphea: Indurated sclerotic plaque; palpable but not visibly elevated (lilac ring at active margin)
- Discoid LE: Follicular plugging, central atrophy, scarring with peripheral hyperpigmentation
5. NODULE
Definition: Palpable, circumscribed, solid, >1 cm involving dermis/subcutis
(See disease-specific images in clinical chapters)
Key nodule diagnoses:
- Erythema nodosum: Tender nodules on anterior shins = septal panniculitis; not ulcerating; causes: Strep, TB, sarcoid, IBD, OCP
- Lipoma: Soft, lobulated, compressible subcutaneous nodule; "slip sign" - slides under fingers
- Dermatofibroma: Firm, indurated nodule; "dimple sign" (Fitzpatrick sign) on lateral pressure; lower leg, women
- Epidermoid cyst: Firm nodule, central punctum, cheesy malodorous contents
- Prurigo nodularis: Hyperkeratotic, crusted nodules from chronic picking; chronic pruritus
6. VESICLE
Definition: Elevated, circumscribed, fluid-filled blister < 1 cm
(Images from previous section available in the full guide)
Key vesicle diagnoses on the ward:
- Herpes zoster: Dermatomal grouped vesicles on erythematous base; unilateral; "zosteriform" distribution
- Herpes simplex: Grouped vesicles, recurrent at same site; fever, lymphadenopathy
- Varicella: "Dew drop on a rose petal" - vesicle on erythematous base, different stages simultaneously, centripetal
- Dyshidrotic eczema (pompholyx): Deep vesicles on palms/soles/lateral fingers; intensely pruritic; summer flare
- Dermatitis herpetiformis: Intensely pruritic vesicles on elbows/knees/buttocks; associated with celiac disease; anti-tTG IgA; granular IgA on DIF
7. BULLA
Definition: Fluid-filled blister > 1 cm
Bulla identification flowchart:Tense bulla (dome-shaped, difficult to break) → Subepidermal → Bullous pemphigoidFlaccid bulla (collapses, breaks easily) → Intraepidermal → Pemphigus vulgaris
Feature Bullous Pemphigoid Pemphigus Vulgaris Blister Tense Flaccid Nikolsky Negative Positive Mucosa Rare Early, prominent Age Elderly (>60) Middle age Level Subepidermal Intraepidermal (suprabasal) Antigen BP180, BP230 Desmoglein 3 (mucosal), Dsg1+3 (mucocutaneous) DIF Linear IgG+C3 at BMZ Intercellular "chicken-wire" IgG
8. PUSTULE
Definition: Elevated, circumscribed, filled with purulent fluid from onset (may be sterile)
Morbilliform drug eruption (macular-papular eruption)
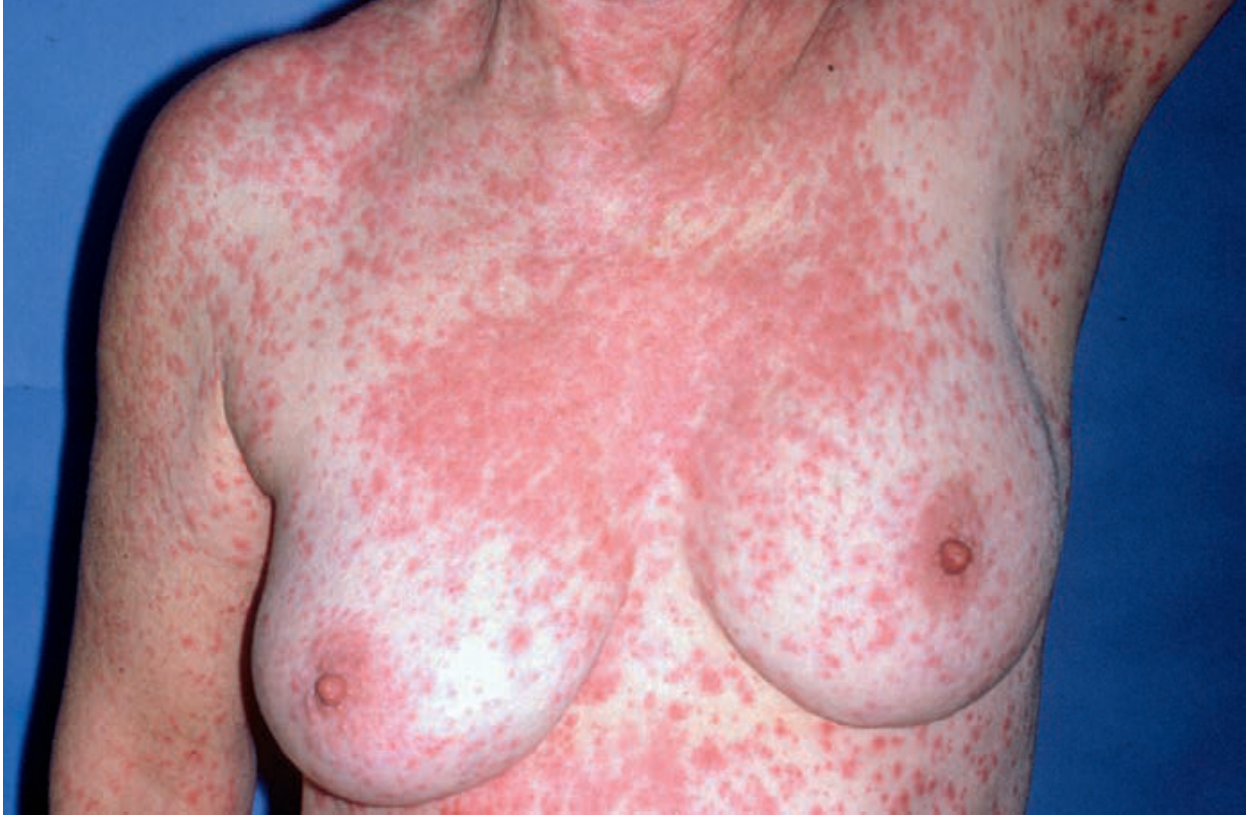 Fig. 2.7 - Generalized morbilliform (measles-like) eruption on trunk. Source: Andrews' Clinical Atlas.
Fig. 2.7 - Generalized morbilliform (measles-like) eruption on trunk. Source: Andrews' Clinical Atlas.
This shows the maculopapular (morbilliform) variant. Note the generalized distribution, blanchable erythema, and confluent pattern. This is the most common drug eruption pattern (aminopenicillins, anticonvulsants, allopurinol).
Pustule diagnoses:
- Follicular pustules: Acne vulgaris (comedones + inflammatory papules/pustules/nodules); folliculitis (Staphylococcus aureus - round pustules around follicle)
- Non-follicular sterile pustules: Pustular psoriasis (GPP - "lakes of pus" on erythematous skin + systemic toxicity; PPP - palmar/plantar); AGEP (acute onset, drug-induced, miliary pustules, fever, rapid resolution)
- Subcorneal pustular dermatosis (Sneddon-Wilkinson): Sterile pustules with hypopyon arrangement; trunk/flexures; older women
- IgA pemphigus: Flaccid pustules, annular configuration; IIF/DIF: intercellular IgA
9. WHEAL
Definition: Transient edematous papule/plaque due to dermal edema; resolves < 24 hours; pruritic
Wheal diagnoses:
- Acute urticaria: Duration <6 weeks; triggers: food (nuts, shellfish), drugs (NSAIDs, penicillin), infection (Strep); IgE-mediated
- Chronic spontaneous urticaria: Duration >6 weeks; no identifiable trigger in >50%; autoimmune (anti-FcεRI, anti-IgE) in ~40%; cyclosporine + omalizumab (anti-IgE) if antihistamines fail
- Dermographism: Linear wheals from stroking; "skin writing"; most common physical urticaria
- Cholinergic urticaria: Small (1-3mm) wheals triggered by heat/exercise/sweating; distinguish from exercise-induced anaphylaxis
REMEMBER: If individual lesion lasts >24h, it may be urticarial vasculitis - order C3/C4, ANA, biopsy (leukocytoclastic infiltrate).
10. CYST
Definition: Encapsulated lesion with true epithelial lining, filled with semi-solid/liquid contents
Cyst diagnoses:
- Epidermoid cyst: Most common; central punctum; cheesy malodorous keratin; face/neck/trunk; may rupture → foreign body granuloma
- Pilar (trichilemmal) cyst: Scalp, familial, may calcify; smoother wall, no granular layer on histology
- Milia: 1-2mm white keratin cysts; face; secondary = healed blisters (EB, PCT - dorsal hands + milia = classic PCT sign)
- Steatocystoma multiplex: Multiple cysts + oily contents; KRT17 mutation; overlaps with pachyonychia congenita
SECONDARY LESIONS
11. SCALE
Definition: Accumulation of stratum corneum (hyperkeratosis)
Scale type → Diagnosis (high-yield for exams):
Scale character Disease Silver/micaceous Psoriasis Powdery/furfuraceous Pityriasis (tinea) versicolor Leading scale (outer edge) Tinea corporis Trailing scale (inner edge) Erythema annulare centrifugum Gritty Actinic keratosis Collarette (peripheral) Pityriasis rosea (secondary lesions) Greasy yellow Seborrheic dermatitis Fish-scale (adherent, limb flexors) X-linked ichthyosis (steroid sulfatase deficiency) Fine, white, generalized Ichthyosis vulgaris (filaggrin mutation) Oyster-shell/rupia crust Crusted (Norwegian) scabies; tertiary syphilis
12. CRUST
Definition: Dried exudate (serum, blood, pus) on skin surface
Crust color → Diagnosis:
Crust color Contents Disease Honey-yellow Serous + bacteria Impetigo (Staph/Strep) - pathognomonic Hemorrhagic Blood Herpes zoster/simplex resolving; vasculitis Yellow-green Pus Secondary infected eczema Oyster-shell (rupia) Layered serous + hemorrhagic Crusted scabies, tertiary syphilis Meliceric Honey-like Bullous impetigo resolving Black eschar Necrotic Anthrax, ecthyma gangrenosum, calciphylaxis
13. EROSION
Definition: Partial (superficial) epidermal loss only → heals WITHOUT scar
Key erosion diagnoses:
- Pemphigus vulgaris (oral erosions appear first; flaccid bullae break → painful erosions)
- Herpes simplex/zoster (vesicles rupture → erosions)
- Any blistering disease post-rupture
- Impetigo (primary = vesicle/bulla → erosion → honey crust)
14. ULCER
Definition: Full-thickness epidermal loss + at least partial dermis → heals WITH scar
Ulcer borders (clinical diagnosis):
Border Disease Violaceous, undermined, "ragged" Pyoderma gangrenosum (pathergy +ve; IBD/RA association) Punched-out, painless Syphilitic chancre (primary syphilis, indurated, clean base) Raised, rolled, pearly BCC (rodent ulcer) Raised, everted, necrotic SCC Sloping, irregular, shallow Venous ulcer (gaiter area, lipodermatosclerosis) Punched-out, exquisitely painful Arterial ulcer (no pulses, pale, pulseless) Painless, callus surrounding Neuropathic ulcer (diabetes, leprosy) Necrotic, rapid onset Ecthyma gangrenosum (Pseudomonas, immunocompromised)
15. LICHENIFICATION
Definition: Accentuation of skin lines due to epidermal thickening from chronic rubbing
- Lichen simplex chronicus: isolated; neck, lower leg, anogenital
- Atopic dermatitis: flexural lichenification (antecubital/popliteal fossae)
- Any chronic pruritic condition
16. ATROPHY
Definition: Acquired loss of skin substance (epidermal = shiny/wrinkled; dermal = depressed)
- Striae distensae: Linear atrophic bands; early = violaceous (striae rubra); late = white (striae alba)
- Anetoderma: Focal dermal atrophy → skin "bulges outward" when pushed; macular atrophy
- Lichen sclerosus: Ivory-white atrophic plaques, anogenital; squamous cell carcinoma risk (~5%)
- Steroid atrophy: Thinning, telangiectasias, striae from prolonged topical steroid use
SPECIAL LESIONS WITH CLINICAL IMAGES
17. CONFIGURATION EXAMPLES
Lichen Planus - flat-topped violaceous papules

Lichen planus diagnosis: 4 P's = Pruritic, Polygonal, Purple (violaceous), Papules. On mucosa: reticular white striae (Wickham striae). Koebner phenomenon +ve. Nail involvement: pterygium (dorsal). Histology: "sawtooth" rete ridges, band-like lymphocytic infiltrate, colloid (Civatte) bodies, hypergranulosis.
Herpes zoster vs Dermatitis herpetiformis (vesicles/bullae)
(Images already fetched from Boliviana block1)
- Herpes zoster: Unilateral, dermatomal vesicles on erythematous base → "zosteriform"
- Dermatitis herpetiformis: Bilateral, extensor surfaces (elbows/knees/buttocks) → "herpetiform" grouping. Both have vesicles but completely different etiology and treatment.
COMPLETE LESION QUICK-REFERENCE WITH DIAGNOSIS
| Lesion | Image available | Key Diagnosis + Distinguishing Feature |
|---|---|---|
| Macule | ✓ Nevus of Ota, Lentigines, Axillary freckling NF1 | Freckle, Vitiligo (depigmented), Café-au-lait (NF1) |
| Patch | ✓ Vitiligo, Nevus depigmentosus | Vitiligo = chalk white (Wood's lamp); Nevus dep = hypopigmented |
| Papule | ✓ Eruptive xanthomas | LP (flat-topped violaceous), Molluscum (umbilicated), Wart (verrucous) |
| Plaque | ✓ (diagram) | Psoriasis (silver scale + Auspitz), MF (indurated), Morphea (indurated, non-elevated) |
| Nodule | - | Erythema nodosum (tender, shins), Dermatofibroma (dimple sign), Lipoma (slip sign) |
| Vesicle | - | HSV (grouped), VZV (dew drop on rose petal), DH (pruritic, extensor) |
| Bulla | - | Tense = BP (subepidermal); Flaccid = PV (intraepidermal, Nikolsky +ve) |
| Pustule | ✓ (drug eruption shown) | GPP (lakes of pus, systemic), AGEP (drug-induced, miliary, rapid resolution) |
| Wheal | - | Urticaria (<24h per lesion), Dermographism (linear), Cholinergic (small, heat-triggered) |
| Cyst | - | Epidermoid (punctum), Pilar (scalp), Milia (1-2mm, white, PCT) |
| Scale | - | Silver = psoriasis; powdery = PV (tinea versicolor); collarette = pityriasis rosea |
| Crust | - | Honey = impetigo; hemorrhagic = HSZ; oyster-shell = crusted scabies |
| Erosion | - | PV (mucosa + skin); heals WITHOUT scar |
| Ulcer | - | Undermined = PG; punched-out = syphilitic chancre; rolled = BCC |
| Lichenification | - | Lichen simplex chronicus; atopic dermatitis flexures |
| Atrophy | - | Striae rubra → alba; anetoderma; lichen sclerosus |
| Telangiectasia | - | Rosacea; CREST (mat); HHT (lips + tongue + fingers) |
| Purpura | - | Non-palpable = thrombocytopenia; palpable = vasculitis (leukocytoclastic) |
| Burrow | - | Scabies (Sarcoptes scabiei); web spaces, wrists, genitalia |
All clinical photographs: Andrews' Clinical Atlas of Skin Disease 2e (©Elsevier) and Dermatology 5e - Boliviana (©Elsevier), reproduced via the institutional medical textbook library.
Posting tip: When you see a new lesion on the ward, immediately ask yourself: (1) Is it flat or raised? → Macule/Patch vs Papule/Plaque/Nodule. (2) Does it have fluid? → Clear fluid = vesicle/bulla; Pus = pustule; Edema = wheal. (3) Is there surface change? → Scale, crust, erosion, lichenification. (4) What is the configuration and distribution? This systematic approach narrows the differential rapidly.
Detail about antifungals
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive content from four authoritative sources. Let me compile the complete guide.
Antifungal Agents - Comprehensive Guide
Sources: Jawetz Medical Microbiology 28e | Andrews' Diseases of the Skin | Dermatology 5e (Boliviana) | Goldman-Cecil Medicine | Medical Microbiology 9e
OVERVIEW: WHY ANTIFUNGALS ARE CHALLENGING
Fungi are eukaryotes, like human cells. Most cellular/molecular processes are similar with extensive gene homology between fungi and mammals - making selective toxicity difficult. Available drug targets must exploit differences between fungal and human cells:
| Target | Difference exploited |
|---|---|
| Ergosterol (cell membrane) | Fungi use ergosterol; humans use cholesterol |
| β-(1,3)-D-glucan (cell wall) | Fungi have cell wall; humans do not |
| Lanosterol 14α-demethylase | Fungal CYP51 is 100-1000× more sensitive to azoles |
| Squalene epoxidase | Fungal enzyme targeted by allylamines |
| Cytosine deaminase | Fungi convert 5-FC to toxic 5-FU; mammalian cells cannot |
| Microtubules | Griseofulvin disrupts fungal mitotic spindle |
CLASSIFICATION BY MECHANISM OF ACTION
ANTIFUNGAL DRUGS
│
├── CELL MEMBRANE (ergosterol binding)
│ └── Polyenes: Amphotericin B, Nystatin
│
├── ERGOSTEROL SYNTHESIS (inhibitors)
│ ├── Azoles (CYP51/14α-demethylase inhibitor)
│ │ ├── Imidazoles: Ketoconazole, Clotrimazole, Miconazole,
│ │ │ Econazole, Oxiconazole, Sertaconazole
│ │ └── Triazoles: Fluconazole, Itraconazole, Voriconazole,
│ │ Posaconazole, Isavuconazole, Otesoconazole
│ ├── Allylamines (squalene epoxidase inhibitor)
│ │ └── Terbinafine, Naftifine, Butenafine
│ └── Others: Amorolfine (morpholine)
│
├── CELL WALL (β-glucan synthesis)
│ └── Echinocandins: Caspofungin, Micafungin, Anidulafungin
│
├── DNA/RNA SYNTHESIS
│ └── Pyrimidine analog: Flucytosine (5-FC)
│
└── MITOSIS (microtubule disruption)
└── Griseofulvin
CLASS 1: POLYENES
Amphotericin B
| Feature | Detail |
|---|---|
| Source | Metabolite of Streptomyces nodosus |
| Mechanism | Binds ergosterol in fungal membrane → forms transmembrane pores → ion/small molecule leakage → membrane disruption |
| Selectivity | Higher affinity for ergosterol (fungal) than cholesterol (mammalian) |
| Action | Fungicidal |
| Route | IV only (poorly absorbed orally) |
| CNS penetration | Poor |
| Resistance | Rare; mechanism = reduced/altered ergosterol (ERG2, ERG3, ERG6 gene defects); inherent resistance in A. terreus, C. auris |
Spectrum: Broad - effective against most major systemic mycoses:
- Candidiasis, Cryptococcosis, Aspergillosis
- Histoplasmosis, Blastomycosis, Coccidioidomycosis
- Mucormycosis (Zygomycetes), Sporotrichosis
Formulations:
| Formulation | Advantage | Toxicity |
|---|---|---|
| Deoxycholate (conventional) AmB | Cheapest | Highest nephrotoxicity |
| Liposomal AmB (L-AmB, AmBisome) | Least nephrotoxic; altered tissue distribution; higher doses possible | Expensive |
| Lipid complex (ABLC, Abelcet) | Less nephrotoxic | Infusion reactions |
| Colloidal dispersion (ABCD) | Less nephrotoxic | Infusion reactions |
Adverse effects (classic "shake and bake"):
- Nephrotoxicity - most important and dose-limiting (renal tubular acidosis, hypokalemia, hypomagnesemia)
- Infusion-related reactions: fever, chills, rigors, headache, hypotension (premedicate with paracetamol, diphenhydramine, hydrocortisone)
- Anemia (reduced erythropoietin)
- Thrombophlebitis at infusion site
- Hypokalemia, hypomagnesemia (electrolyte monitoring essential)
Uses:
- Empiric therapy in febrile neutropenia when unresponsive to antibacterials
- Severe/life-threatening invasive mycoses (when azoles not suitable)
- Mucormycosis (echinocandins NOT active)
- Cryptococcal meningitis induction (L-AmB + flucytosine × 2 weeks)
Nystatin
| Feature | Detail |
|---|---|
| Mechanism | Same as amphotericin B - binds ergosterol; pore formation |
| Route | Topical only (too toxic for systemic use) |
| Absorption | No systemic absorption |
| Use | Oral/cutaneous/vaginal candidiasis ONLY |
| NOT effective against | Dermatophytes |
| Side effects | None (no systemic absorption) |
Formulations: Oral suspension ("swish and swallow"), lozenges/pastilles, cream, ointment, vaginal tablets
CLASS 2: AZOLES
Mechanism (All Azoles)
Block CYP51 (lanosterol 14α-demethylase) - a cytochrome P450 enzyme - preventing conversion of lanosterol to ergosterol. This depletes ergosterol and causes accumulation of methylated sterols → increased membrane permeability, disrupted enzyme activity, inhibition of fungal growth.
Action: Primarily fungistatic (fungicidal at high concentrations for some species)
Drug interactions (critical): All azoles inhibit mammalian CYP enzymes (especially CYP3A4, CYP2C9):
- Cyclosporine, tacrolimus, sirolimus → increased levels → toxicity
- Warfarin → increased anticoagulation
- Statins (simvastatin, lovastatin) → increased levels → myopathy
- Phenytoin, rifampicin → decreased azole levels
- Antacids, H2 blockers, PPIs → decreased ketoconazole/itraconazole absorption
IMIDAZOLES (2 nitrogens in ring)
Ketoconazole
| Feature | Detail |
|---|---|
| Route | Oral, topical |
| Spectrum | Dermatophytes, Candida, some endemic mycoses |
| Absorption | Requires acidic pH; reduced by antacids/PPIs/H2 blockers |
| Special effect | Inhibits testosterone and cortisol synthesis → gynecomastia, decreased libido, adrenal insufficiency |
| Hepatotoxicity | Significant; most toxic of azoles |
| FDA warning | Should NOT be used for skin/nail infections (systemic form); only for systemic mycoses when NO other option |
| Topical use | Still widely used: 2% cream/gel, 1-2% shampoo for seborrheic dermatitis/tinea versicolor |
Topical Imidazoles (all available OTC or Rx)
| Drug | Formulation | Notes |
|---|---|---|
| Clotrimazole | 1% cream/lotion/solution, lozenges, vaginal | Broad-spectrum; oral troches for oropharyngeal candidiasis |
| Miconazole | 2% cream/powder/spray, vaginal | OTC; broad-spectrum |
| Econazole | 1% cream | Rx; good for tinea pedis |
| Oxiconazole | 1% cream/lotion | Rx |
| Sertaconazole | 2% cream | Rx; anti-inflammatory + antifungal |
| Luliconazole | 1% cream | Rx; excellent nail/skin penetration |
| Butoconazole | 2% vaginal cream | OTC; Candida vaginitis |
| Ticonazole | 6.5% vaginal cream, 20% nail lacquer | OTC |
TRIAZOLES (3 nitrogens in ring)
Fluconazole
| Feature | Detail |
|---|---|
| Route | Oral (high bioavailability, nearly 100%), IV |
| Spectrum | Candida spp., Cryptococcus neoformans; limited mold activity |
| CNS penetration | Excellent (most hydrophilic azole) → used for fungal meningitis |
| Absorption | NOT affected by gastric acid or food |
| Half-life | ~30 hours (allows once-daily or weekly dosing) |
| Resistance | Inherent in C. krusei; frequently acquired in C. glabrata |
| Side effects | Generally mild: GI upset, hepatotoxicity (rare), Stevens-Johnson (rare), QTc prolongation |
| Drug of choice | Systemic candidiasis (non-neutropenic); cryptococcal meningitis consolidation/maintenance; oropharyngeal/vaginal candidiasis |
Dosing (Dermatology 5e - Boliviana):
| Indication | Adult Dose |
|---|---|
| Vaginal candidiasis | 150 mg single dose |
| Oropharyngeal candidiasis | 200 mg day 1, then 100 mg/day × 2 weeks |
| Tinea corporis/cruris | 50-100 mg/day OR 150 mg weekly × 2-4 weeks |
| Tinea pedis | 150-450 mg weekly × 2-6 weeks |
| Tinea versicolor | 400 mg once OR 300 mg once (repeat in 1 week) |
| Onychomycosis | 150-450 mg weekly × 6 mo (fingernails) or 9 mo (toenails) |
| Systemic candidiasis | 800 mg day 1, then up to 400 mg/day |
| Cryptococcal meningitis consolidation | 400 mg/day × 8 weeks |
| Cryptococcal meningitis maintenance | 200 mg/day × 6-12 months |
| Prevention of relapse (HIV) | 200 mg daily |
Itraconazole
| Feature | Detail |
|---|---|
| Route | Oral (capsules - poor/variable absorption; solution - better absorption), IV |
| Spectrum | Broad: dermatophytes, Candida, Aspergillus, endemic mycoses, Sporothrix |
| Absorption | Capsules: requires fatty food and acid pH; Solution: better absorbed but causes diarrhea |
| Side effects | Hepatotoxicity, negative inotrope (avoid in cardiac failure), GI upset, neuropathy, hypokalemia |
| Drug of choice | Histoplasmosis (non-CNS), Blastomycosis, Paracoccidioidomycosis, onychomycosis |
Dosing (Dermatology 5e - Boliviana):
| Indication | Dose |
|---|---|
| Tinea corporis/cruris | 100 mg/day × 2 weeks OR 200 mg/day × 1 week |
| Tinea pedis | 100 mg/day × 2 weeks OR 200 mg/day × 1 week |
| Tinea versicolor | 200 mg/day × 5-7 days |
| Onychomycosis toenail (continuous) | 200 mg/day × 12 weeks |
| Onychomycosis toenail (pulse) | 200 mg BID × 1 week, off 3 weeks; repeat ×3 |
| Onychomycosis fingernail (pulse) | 200 mg BID × 1 week, off 3 weeks; repeat ×1 (2 pulses) |
| Aspergillosis | 200-400 mg/day; loading 200 mg TID × 3 days if severe |
| Histoplasmosis/Blastomycosis | 200 mg/day (may increase to 400 mg/day) × ≥3 months |
| Oropharyngeal candidiasis | 200 mg/day swish and swallow × 1-2 weeks |
| Esophageal candidiasis | 100-200 mg/day |
Voriconazole
| Feature | Detail |
|---|---|
| Route | Oral (excellent absorption), IV |
| Spectrum | Broadest of triazoles: Aspergillus, Candida (including non-albicans), Fusarium, Scedosporium, Cryptococcus, endemic fungi, many rare molds |
| NOT active against | Mucorales (Zygomycetes) |
| Drug of choice | Invasive aspergillosis (first-line); Fusariosis; Scedosporiosis |
| CNS penetration | Good |
| Visual side effects | Reversible visual disturbances (photopsia, blurred vision, color changes) in ~30% |
| Dermatology-specific ADRs | Photosensitivity, premature photoaging, actinic keratoses, squamous cell carcinoma, melanoma, porphyria-like reactions, drug-induced lentigines |
| Metabolized by | CYP2C19 (genetically variable) → therapeutic drug monitoring recommended |
| Contraindications | Elevated liver enzymes, prolonged QTc, history of intolerance |
Posaconazole
| Feature | Detail |
|---|---|
| Route | Oral (suspension, delayed-release tablet), IV |
| Spectrum | Broadest: includes Mucorales (Zygomycetes) |
| Drug of choice | Mucormycosis (with L-AmB); prophylaxis in high-risk neutropenic patients |
| Advantage over voriconazole | Active against Mucorales; fewer cutaneous/visual ADRs |
| Side effects | Generally well tolerated; GI upset, hepatotoxicity, QTc prolongation |
| Monitoring | Serum levels required (absorption is highly variable) |
Isavuconazole (Isavuconazonium sulfate = prodrug)
| Feature | Detail |
|---|---|
| Route | Oral, IV |
| Approved for | Invasive aspergillosis, Mucormycosis |
| Advantages | More predictable pharmacokinetics, fewer drug-related ADRs vs voriconazole; NO QTc prolongation (actually shortens QTc) |
| Loading dose | 372 mg q8h × 6 doses, then 372 mg once daily |
| Status | Increasingly becoming drug of choice for aspergillosis (especially in patients with QTc concerns or liver disease) |
Otesoconazole (newest)
| Feature | Detail |
|---|---|
| Route | Oral |
| Approved for | Recurrent vulvovaginal candidiasis in women without reproductive potential |
| Advantage | Very long half-life; minimal CYP inhibition compared to other azoles |
CLASS 3: ALLYLAMINES
Terbinafine
| Feature | Detail |
|---|---|
| Mechanism | Inhibits squalene epoxidase → blocks conversion of squalene to lanosterol → accumulation of squalene (toxic to fungi) + depletion of ergosterol |
| Action | Fungicidal against dermatophytes (unlike azoles which are fungistatic) |
| Route | Oral, topical (1% cream/gel) |
| Spectrum | Dermatophytes (excellent), some Candida, limited Malassezia |
| Weakness | Less active against Candida and Microsporum in vitro (though adequate doses effective in vivo); limited efficacy against tinea versicolor orally |
| Accumulates in | Stratum corneum, hair, nails (keratin-binding = sustained activity after discontinuation) |
| Half-life | Very long (300+ hours in nails) |
| Hepatotoxicity | Rare but possible - LFT monitoring if prolonged |
| Taste disturbance | Ageusia/dysgeusia - infrequent but troublesome |
| Other ADRs | GI distress, headache, skin reactions, leukopenia, TEN (rare) |
| Drug interactions | Less than azoles; bioavailability unchanged with food |
Drug of choice: Dermatophyte infections, especially onychomycosis (higher cure rate than itraconazole - 70% toenail, 80% fingernail)
Dosing:
- Onychomycosis toenail: 250 mg/day × 12 weeks
- Onychomycosis fingernail: 250 mg/day × 6 weeks
- Tinea capitis: 250 mg/day × 2-6 weeks (for Trichophyton spp.; less effective for Microsporum)
- Superficial tinea: 250 mg/day × 2 weeks (or topical 1-2 weeks)
Naftifine & Butenafine
- Topical allylamines
- Used for tinea pedis, tinea cruris, tinea corporis
- Naftifine: 1% cream/gel; Butenafine: 1% cream (classified as benzylamine - similar MOA)
CLASS 4: ECHINOCANDINS
Caspofungin, Micafungin, Anidulafungin
| Feature | Detail |
|---|---|
| Mechanism | Non-competitive inhibition of β-(1,3)-D-glucan synthase → disrupts fungal cell wall synthesis → osmotic instability and cell lysis |
| Action | Fungicidal against Candida; Fungistatic against Aspergillus |
| Route | IV only (not orally absorbed) |
| Spectrum | Most Candida spp. (including azole-resistant), Aspergillus spp. |
| NOT active against | Mucorales, Cryptococcus neoformans, Fusarium, Trichosporon |
| Resistance | Rare; mutations in FKS1/FKS2 (β-glucan synthase genes); C. auris may have primary resistance |
| Metabolism | Hepatic (non-CYP) → fewer drug interactions than azoles |
| Renal dosing | No dose adjustment needed for renal impairment |
| Side effects | Generally well tolerated: phlebitis, fever, elevated liver enzymes, mild hemolysis, histamine-like infusion reaction |
Specific approvals:
| Drug | Key Indications |
|---|---|
| Caspofungin | Invasive aspergillosis (refractory/intolerant); invasive/esophageal candidiasis; empiric therapy in febrile neutropenia |
| Micafungin | Candidiasis; prophylaxis in HSCT recipients; invasive aspergillosis (second-line) |
| Anidulafungin | Candidemia, esophageal candidiasis; combination with voriconazole for invasive aspergillosis (reduces mortality) |
Key clinical use:
- Preferred over fluconazole in critically ill/septic patients with candidiasis
- Activity in azole-resistant Candida (C. glabrata, C. krusei)
- Can be combined with voriconazole or L-AmB for refractory infections
CLASS 5: PYRIMIDINE ANALOGUE
Flucytosine (5-FC, 5-Fluorocytosine)
| Feature | Detail |
|---|---|
| Mechanism | Transported into fungal cells by cytosine permease → deaminated by fungal cytosine deaminase to 5-fluorouracil (5-FU) → incorporated as 5-fluorooxyuridylic acid → inhibits thymidylate synthase and DNA synthesis. Mammalian cells lack cytosine deaminase → selective toxicity |
| Route | Oral |
| Spectrum | Candida, Cryptococcus, Chromoblastomycosis (Dematiaceous molds) |
| CNS penetration | Excellent |
| Used as | Almost ALWAYS in combination with amphotericin B (never monotherapy) |
| Resistance | Emerges rapidly if used alone → avoid monotherapy |
| Synergy | Synergistic with amphotericin B (ampB increases cell permeability → more 5-FC enters); also delays resistance emergence |
| Side effects | Bone marrow suppression (anemia, leukopenia, thrombocytopenia), GI disturbance (colitis - from intestinal bacterial conversion), hepatotoxicity; AIDS patients especially susceptible to bone marrow suppression |
| Monitoring | Serum levels essential (narrow therapeutic window); renal function (cleared renally - dose adjust in renal impairment) |
Standard use: L-AmB + flucytosine × 2 weeks induction → fluconazole consolidation, in Cryptococcal meningitis
CLASS 6: GRISEOFULVIN
| Feature | Detail |
|---|---|
| Source | Derived from Penicillium griseofulvum |
| Mechanism | Binds tubulin → disrupts mitotic spindle formation → inhibits mitosis of fungal hyphae. Only affects actively growing hyphae. |
| Route | Oral only |
| Spectrum | Dermatophytes only (no effect on yeast or other fungi) |
| Distribution | Concentrates in keratinized tissues (stratum corneum, hair, nails) |
| Absorption | Poor and variable; increased by fatty food; microsize/ultramicrosize formulations improve absorption |
| FDA approved | Tinea capitis in children >2 years (drug of choice in pediatric tinea capitis - especially for Microsporum spp.) |
| Duration | Long courses required (weeks to months) |
| Side effects | Headache (most common, resolves spontaneously), GI disturbance, drowsiness, photosensitivity, hepatotoxicity (rare), teratogenicity (contraindicated in pregnancy), lupus-like syndrome |
| Drug interactions | Induces CYP450 → reduces levels of warfarin, OCP, cyclosporine; alcohol → disulfiram-like reaction |
Dosing:
- Children: 10-20 mg/kg/day (microsize) or 5-10 mg/kg/day (ultramicrosize)
- Adults: 500 mg/day (microsize) or 330-375 mg/day (ultramicrosize)
- Tinea capitis: 6-12 weeks
- Tinea unguium (nails): 6-12 months (fingernails) or 12-18 months (toenails)
Key exam point: Griseofulvin remains drug of choice for tinea capitis caused by Microsporum spp. in children (terbinafine is preferred for Trichophyton spp. tinea capitis and is FDA approved from age 4).
TOPICAL ANTIFUNGALS: ADDITIONAL AGENTS
Ciclopirox Olamine
- Unique mechanism: Chelates polyvalent metal cations (Fe3+, Al3+) → inhibits metal-dependent enzymes → disrupts multiple fungal pathways (cell membrane, mitochondria, DNA repair)
- Spectrum: Broad - dermatophytes, Candida, Malassezia; also anti-inflammatory and antibacterial
- Use: Tinea pedis/corporis/cruris/versicolor, onychomycosis (8% nail lacquer × 48 weeks), seborrheic dermatitis (1% shampoo)
- Nail lacquer: Apply daily × 1 week, clean with alcohol; repeat cycle × 48 weeks
Tolnaftate
- Mechanism: Inhibits squalene epoxidase (like allylamines)
- Use: Dermatophytes, Malassezia; NOT active against Candida
- OTC available; less effective than azoles or allylamines
Undecylenic Acid
- Antifungal fatty acid; OTC; mainly for athlete's foot
Amorolfine (morpholine)
- Topical nail lacquer; inhibits sterol synthesis at two sites (different from azoles)
- Used for onychomycosis; available in Europe/Asia
Tavaborole
- Boron-containing antifungal (inhibits leucyl-tRNA synthetase)
- Topical 5% solution for onychomycosis; applied daily × 48 weeks
- Advantage: smaller molecule → better nail penetration
Efinaconazole
- Triazole nail lacquer (10% solution)
- Applied daily × 48 weeks for onychomycosis
- Good nail plate penetration; low protein binding in nail
DRUG RESISTANCE MECHANISMS
| Drug Class | Mechanism of Resistance |
|---|---|
| Azoles | 1. ERG11 mutation (target alteration - reduced azole binding) 2. CDR1/CDR2 efflux pumps (ABC transporters) 3. MDR1 efflux pump (MFS transporter) 4. ERG3 mutation (bypasses need for ergosterol) 5. Overexpression of ERG11 |
| Polyenes | 1. Reduced ergosterol content (ERG2, ERG3, ERG6 mutations) 2. Replacement of ergosterol by less-binding sterols (fecosterol) 3. Masking of ergosterol |
| Echinocandins | FKS1/FKS2 hotspot mutations (β-glucan synthase gene) |
| Flucytosine | 1. Loss of cytosine permease (transport) 2. Loss of UPRTase (incorporation enzyme) → reduced conversion of 5-FC |
| Note | Fungi CANNOT destroy/modify antifungal drugs (unlike bacteria). No horizontal gene transfer of resistance. Resistance develops slowly by stepwise alteration. |
Inherently resistant species (key exam facts):
- C. krusei → inherently resistant to fluconazole
- C. glabrata → frequently acquired resistance to fluconazole
- C. auris → often multidrug resistant (fluconazole, amphotericin B)
- A. terreus → inherently resistant to amphotericin B
- Mucorales → resistant to echinocandins and voriconazole
ANTIFUNGAL SELECTION BY INFECTION TYPE
| Infection | First-Line | Alternative |
|---|---|---|
| Tinea capitis (Trichophyton) | Terbinafine | Fluconazole, Itraconazole |
| Tinea capitis (Microsporum) | Griseofulvin | Fluconazole |
| Tinea corporis/cruris/pedis | Topical azole or terbinafine | Oral terbinafine or itraconazole (if extensive) |
| Tinea versicolor | Topical selenium sulfide/ketoconazole shampoo | Oral itraconazole 200 mg/d × 5-7d or fluconazole 400 mg once |
| Onychomycosis | Terbinafine 250 mg/d × 12 wk (toenail) | Itraconazole pulse × 3 pulses; topical (nail lacquer) if limited |
| Oropharyngeal candidiasis | Fluconazole 200 mg day 1, 100 mg/d × 14d | Nystatin suspension; itraconazole |
| Vulvovaginal candidiasis | Fluconazole 150 mg single dose | Topical azole × 1-7 days |
| Candidiasis (non-neutropenic ICU) | Echinocandin (caspofungin, micafungin) | Fluconazole (if not critically ill, stable, no azole exposure) |
| Invasive Aspergillosis | Voriconazole OR Isavuconazole | Liposomal AmB; Posaconazole; +/- echinocandin combination |
| Mucormycosis | Liposomal AmB + Posaconazole | Isavuconazole |
| Cryptococcal meningitis (induction) | Liposomal AmB 3-4 mg/kg/d + Flucytosine 25 mg/kg QID × 2 weeks | Conventional AmB + 5-FC |
| Cryptococcal meningitis (consolidation) | Fluconazole 400 mg/d × 8 weeks | Itraconazole |
| Cryptococcal meningitis (maintenance/HIV) | Fluconazole 200 mg/d × 6-12 months | - |
| Histoplasmosis/Blastomycosis (mild-mod) | Itraconazole | - |
| Histoplasmosis (severe/CNS) | Liposomal AmB | - |
| Febrile neutropenia (empiric) | Echinocandin OR L-AmB | Voriconazole if mold suspected |
| Prophylaxis (HSCT/AML) | Fluconazole OR Posaconazole | Micafungin, voriconazole |
SUMMARY COMPARISON TABLE
| Drug | Class | MOA | Action | Route | Spectrum | Key ADR | Drug of Choice |
|---|---|---|---|---|---|---|---|
| Amphotericin B | Polyene | Ergosterol binding (membrane) | Cidal | IV | Broadest | Nephrotoxicity, infusion reactions | Severe systemic mycoses, Mucormycosis |
| Nystatin | Polyene | Same | Cidal | Topical | Candida only | None | Oral/cutaneous/vaginal candidiasis |
| Fluconazole | Triazole | CYP51 inhibition | Static | PO/IV | Candida, Crypto | Mild GI, CYP interactions | Candidiasis, Crypto maintenance |
| Itraconazole | Triazole | CYP51 inhibition | Static | PO | Broad + endemic | Cardiac failure, hepatotoxicity | Histoplasma, Blastomyces, onycho |
| Voriconazole | Triazole | CYP51 inhibition | Static | PO/IV | Broadest triazole | Visual, photosensitivity, SCC | Invasive Aspergillosis |
| Posaconazole | Triazole | CYP51 inhibition | Static | PO | Broadest (+ Mucor) | GI, level monitoring | Mucormycosis, prophylaxis |
| Isavuconazole | Triazole | CYP51 inhibition | Static | PO/IV | Broad | Least ADRs | Aspergillosis (QTc issues), Mucor |
| Terbinafine | Allylamine | Squalene epoxidase | Cidal | PO/topical | Dermatophytes | Taste loss, hepatotoxicity | Onychomycosis, tinea capitis (T.) |
| Griseofulvin | - | Microtubule disruption | Static | PO | Dermatophytes ONLY | Headache, photosensitivity | Tinea capitis (Microsporum), children |
| Flucytosine | Pyrimidine | DNA/RNA synthesis | Cidal (combo) | PO | Candida, Crypto | Bone marrow suppression | Combo with AmB for Crypto meningitis |
| Caspofungin | Echinocandin | β-glucan synthase | Cidal (Candida) | IV | Candida, Aspergillus | Well tolerated | Candidiasis (ICU), refractory Aspergillus |
| Micafungin | Echinocandin | β-glucan synthase | Cidal (Candida) | IV | Candida, Aspergillus | Well tolerated | Candidiasis, HSCT prophylaxis |
| Anidulafungin | Echinocandin | β-glucan synthase | Cidal (Candida) | IV | Candida, Aspergillus | Well tolerated | Candidiasis + combo for Aspergillus |
| Ciclopirox | Miscellaneous | Metal chelation | Static/cidal | Topical | Broad | None significant | Onychomycosis (nail lacquer), tinea |
Sources: Jawetz Melnick & Adelberg's Medical Microbiology 28e, Ch. 45 | Andrews' Diseases of the Skin, Ch. 15 | Dermatology 5e (Boliviana), Ch. 127 | Goldman-Cecil Medicine, Ch. 311 | Medical Microbiology 9e, Ch. 61
Hereditary angioedema detail along with biochemistry
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the information needed for a comprehensive PG-level HAE guide. Let me compile it.
Hereditary Angioedema (HAE) - PG Dermatology Guide
1. Overview and Historical Note
HAE was originally described and named by William Osler in 1888 as "Quincke edema." It is a potentially life-threatening condition affecting approximately 1 in 50,000-150,000 persons, characterized by recurrent, self-limited episodes of non-pitting, non-pruritic angioedema.
Key clinical hallmarks that distinguish HAE from allergic angioedema:
- NO urticaria or pruritus
- NO response to antihistamines, epinephrine, or corticosteroids
- Frequent GI involvement (can mimic surgical emergency)
- Laryngeal edema risk (main cause of mortality)
- Duration: 48-72 hours per attack, often followed by a refractory period
2. Classification of HAE - Three Types
| Feature | Type I | Type II | Type III (nC1-INH-HAE) |
|---|---|---|---|
| C1-INH antigen | LOW (↓↓) | Normal or HIGH | Normal |
| C1-INH function | Low | LOW (dysfunctional) | Normal |
| C4 level | Always LOW | Always LOW | Normal |
| C1q level | Normal | Normal | Normal |
| Gene | SERPING1 mutation | SERPING1 mutation | F12, PLG, ANGPT1, KNG1, MYOF, HS3ST6 |
| Inheritance | Autosomal dominant | Autosomal dominant | AD (gain-of-function) |
| Proportion | 85% of cases | 15% of cases | Rare |
| Sex predominance | M = F | M = F | Predominantly women |
Type III is now further subdivided into HAE3-HAE8 based on the mutated gene - HAE3 (F12 mutation), HAE4 (PLG/plasminogen), HAE5 (ANGPT1/angiopoietin-1), HAE6 (KNG1/kininogen-1), HAE7 (MYOF/myoferlin), HAE8 (HS3ST6); a 7th subgroup with unknown mutation also exists.
3. Genetics and Molecular Basis
Type I & II: Heterozygous loss-of-function mutation in ONE allele of SERPING1 gene (chromosome 11q11-q13.1), which encodes C1 esterase inhibitor (C1-INH / C1-INH), a serine protease inhibitor (serpin).
- Expected level with one non-functional allele = 50% of normal
- Actual measured level = only 5-30% of normal (type I)
- Reason: trans-inhibition of the normal allele OR increased catabolism of C1-INH
- Rare homozygous autosomal recessive severe HAE also reported
Type III (HAE3): Gain-of-function mutations in F12 (Factor XII gene) → increased kallikrein formation → excess bradykinin. Estrogens upregulate F12 transcription - explains female predominance and exacerbation by oral contraceptives.
4. Biochemistry - The Kallikrein-Kinin System
This is the central biochemical cascade. Understanding it is essential for understanding both HAE pathogenesis and drug targets.
4a. Normal Kinin Pathway
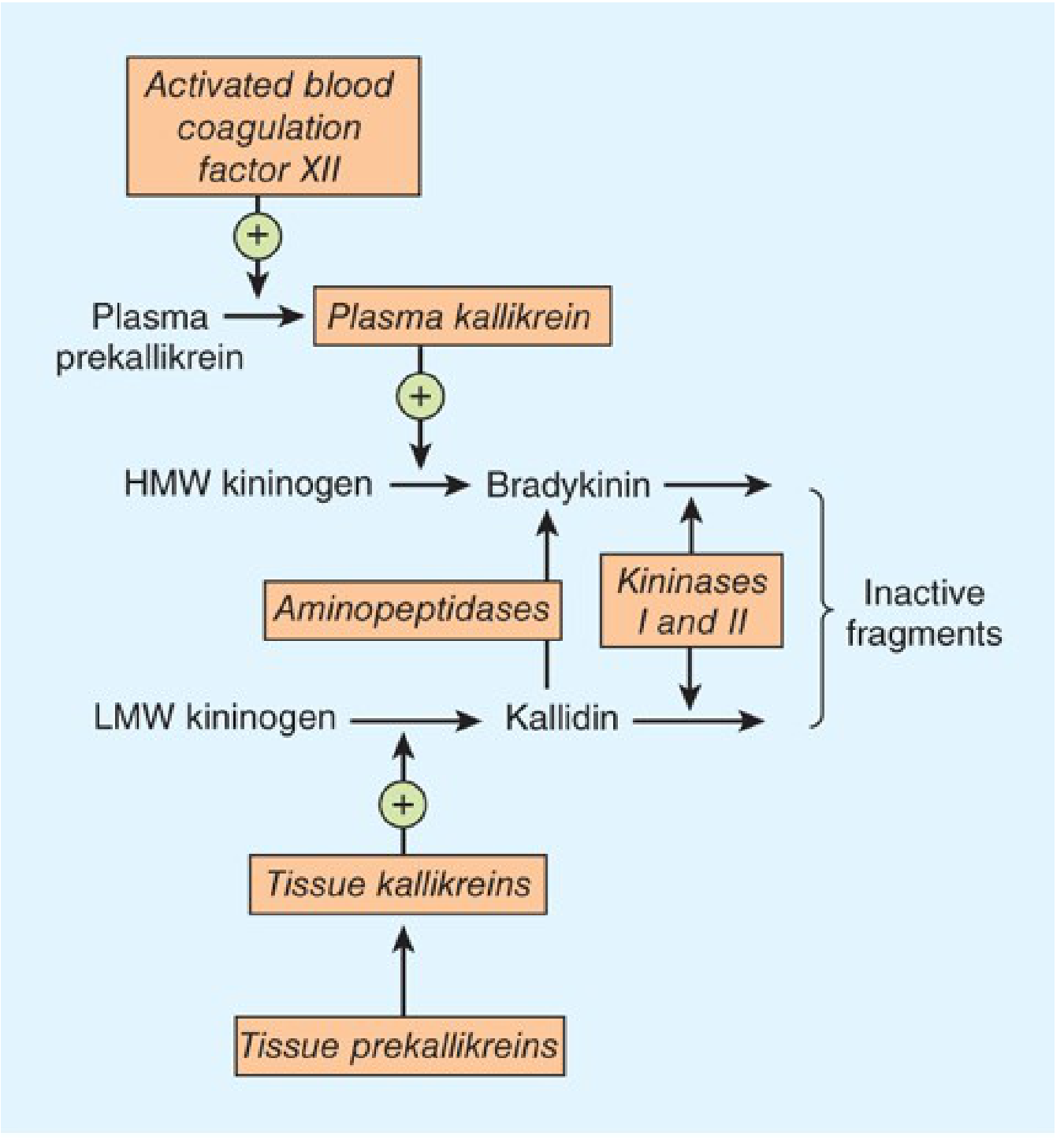
Key components:
- Kallikreins - serine proteases existing in two forms:
- Plasma kallikrein - activated by FXIIa (Hageman factor); cleaves HMW kininogen → bradykinin (9 amino acids)
- Tissue kallikrein - cleaves LMW kininogen → kallidin (Lys-bradykinin, 10 amino acids)
- Kininogens - precursor proteins, synthesized in liver:
- HMW kininogen - confined to bloodstream, substrate for plasma kallikrein
- LMW kininogen - crosses capillary walls, substrate for tissue kallikrein
- Kinins - potent vasoactive peptides (half-life <15 seconds):
- Bradykinin = predominant plasma kinin
- Kallidin = major urinary form; can be converted to bradykinin by aminopeptidase
- Kinin degradation (kininases):
- Kininase I (carboxypeptidase N) - removes C-terminal arginine from bradykinin
- Kininase II = ACE (angiotensin-converting enzyme) - the same enzyme! Converts angiotensin I → angiotensin II AND inactivates bradykinin
Exam pearl: This is why ACE inhibitors cause bradykinin-mediated angioedema - they block kininase II, preventing bradykinin degradation.
4b. Bradykinin Receptor Pharmacology
| Receptor | Expression | Key Functions |
|---|---|---|
| B2 receptor | Constitutive, widespread | Mediates most acute effects of bradykinin - vasodilation, increased vascular permeability, pain, bronchoconstriction |
| B1 receptor | Inducible (upregulated by tissue injury, infection, inflammation) | Chronic pain, inflammatory disease (vasculitis, neuroinflammation) |
Both are GPCRs → activate PLC → IP3/DAG → Ca²+ mobilization + NO + prostaglandins + MAPK activation.
4c. C1-INH and Its Substrates
C1-INH is a serpin (serine protease inhibitor) that physiologically suppresses:
| System | Enzymes Inhibited |
|---|---|
| Complement | C1r, C1s (classical pathway initiation) |
| Contact/Kinin | Plasma kallikrein, Factor XIIa (FXIIa, Hageman factor) |
| Coagulation | Factor XIa |
| Fibrinolysis | Plasmin (partially) |
By simultaneously controlling all four systems, C1-INH is a master inhibitor at the interface of innate immunity and coagulation.
5. Pathophysiology of HAE - The Central Biochemical Loop
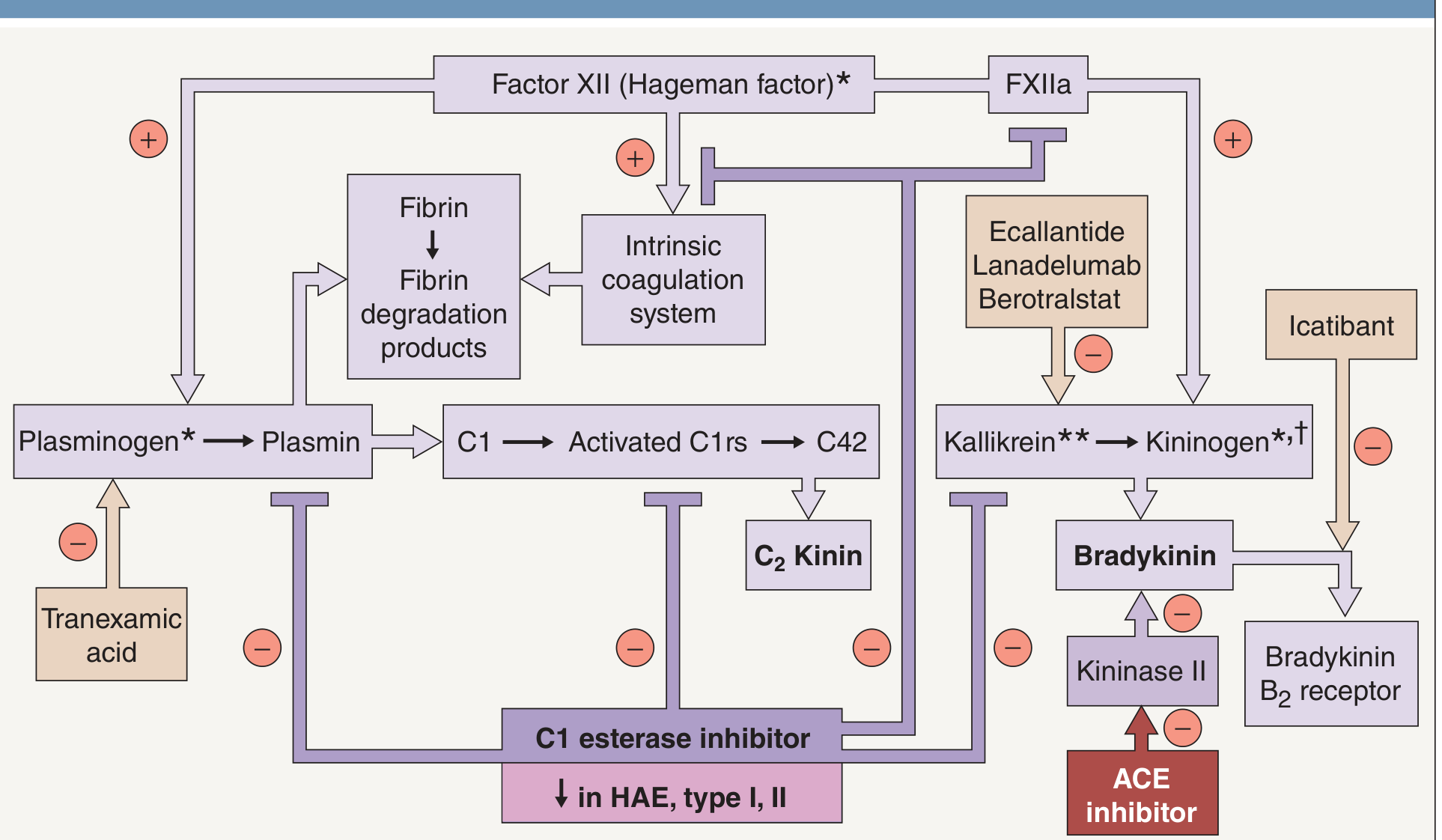
Step-by-step cascade in HAE (Type I/II):
Trigger (trauma/surgery/estrogen/emotional stress)
↓
Factor XII (FXII) activation → FXIIa
↓
FXIIa activates Plasma prekallikrein → Plasma KALLIKREIN
↓
Kallikrein cleaves HMW kininogen → BRADYKININ ↑↑↑
↓
(C1-INH absent/dysfunctional - no brake!)
↓
BRADYKININ → B2 receptor on endothelium
↓
NO + PGI2 + substance P
↓
↑↑ Vascular permeability → ANGIOEDEMA
Simultaneously:
Plasmin activates C1 → C1r, C1s active → C4 and C2 cleaved/consumed
↓
C4 perpetually LOW (even between attacks) ← DIAGNOSTIC HALLMARK
↓
C2 kinin also generated (contributes to swelling)
Positive feedback loop: Kallikrein also activates more FXII → amplification cycle.
Why NO urticaria? Bradykinin does NOT degranulate mast cells. Histamine plays NO role. Hence antihistamines are completely ineffective.
6. Complement Profile - Diagnostic Biochemistry
| Parameter | Type I HAE | Type II HAE | Type III HAE | Acquired C1-INH deficiency | ACE inhibitor-induced |
|---|---|---|---|---|---|
| C1-INH antigen | ↓↓↓ | Normal or ↑ | Normal | ↓ | Normal |
| C1-INH function | ↓ | ↓↓ (dysfunctional) | Normal | ↓ | Normal |
| C4 | ↓↓ (always, even between attacks) | ↓↓ | Normal | ↓ | Normal |
| C2 | ↓ during attacks | ↓ during attacks | Normal | ↓ | Normal |
| C1q | Normal | Normal | Normal | ↓ (KEY) | Normal |
| C3 | Normal | Normal | Normal | Normal or ↓ | Normal |
| C1 function (CH50) | ↓ | ↓ | Normal | ↓ | Normal |
Key diagnostic tip: In Type I/II HAE, C1q is NORMAL. In Acquired C1-INH deficiency, C1q is LOW (because anti-C1q antibodies or malignancy consumes C1q along with the whole C1 complex). This C1q level is what differentiates acquired from hereditary.
Screening test of choice for Type I/II = C4 level. C4 is chronically low (<40% of normal) due to constant activation and consumption - even between attacks, at rest.
Lab during an attack:
- Urinary histamine elevated
- Serum C1 levels elevated
- CH50, C4, C2 all decrease further
7. Triggers of HAE Attacks
- Physical trauma (minor cuts, dental procedures)
- Surgery/intubation (major risk period)
- Emotional stress
- Estrogens - oral contraceptives, pregnancy (upregulate F12 transcription and bradykinin sensitivity)
- ACE inhibitors (compound the defect by reducing kininase II activity)
- Infections
- Sudden temperature changes
- Prodromal sign: some families develop a transient erythema marginatum-like rash before an attack (NOT urticaria)
8. Clinical Features
Sites of involvement:
| Site | Manifestation | Risk |
|---|---|---|
| Skin (subcutaneous) | Face, hands, feet, genitals, buttocks - non-pitting, asymmetric, non-pruritic | Low mortality |
| GI (submucosal) | Nausea, vomiting, severe colic, abdominal pain | Misdiagnosis as surgical emergency; unnecessary appendectomy |
| Larynx/upper airway | Hoarseness → stridor → asphyxia | Life-threatening - main cause of death |
| Urogenital | Bladder, genital swelling | Uncomfortable |
Key features:
- Onset usually before age 20 (2nd decade most common)
- Attacks last 48-72 hours (range: 2-5 days)
- Frequency: as often as every 2 weeks
- Between attacks: C4 always low, but patient is asymptomatic
- ABSENT: urticaria, pruritus, flushing, bronchospasm
9. Diagnosis - Algorithm
Recurrent angioedema WITHOUT urticaria/pruritus
↓
Check C4 level
↓ LOW → Check C1-INH antigen + C1-INH function
↓
C1-INH antigen LOW + function LOW → Type I HAE
C1-INH antigen normal/HIGH + function LOW → Type II HAE
C1q also LOW? → Acquired C1-INH deficiency
↓ NORMAL C4 → Check FXII mutation, PLG, KNG1, ANGPT1
→ Type III HAE (nC1-INH-HAE)
Also check:
- Family history (AD pattern - 50% offspring affected)
- Age of onset (HAE typically <20 years; acquired typically >40 years)
- Response to treatment (HAE doesn't respond to antihistamines/steroids)
- SERPING1 genetic sequencing for confirmation
- In acquired form: look for lymphoproliferative disease, monoclonal gammopathy, SLE
10. Treatment - Comprehensive Pharmacology
10a. Acute Attack Treatment
| Drug | Class | Mechanism | Route | Dosing | Notes |
|---|---|---|---|---|---|
| Plasma-derived C1-INH (Berinert, Cinryze) | Replacement therapy | Directly replaces deficient C1-INH | IV | 20 IU/kg | First-line for acute attacks |
| Recombinant C1-INH (Ruconest/Conestat alfa) | Replacement therapy | Derived from transgenic rabbit milk | IV | 50 IU/kg | Same MOA, lower blood product risk |
| Icatibant (Firazyr) | Bradykinin B2 receptor antagonist | Competitive, highly selective B2 blocker | SC | 30 mg SC, can repeat ×2 every 6h | Decapeptide; also useful in ACE inhibitor-induced angioedema |
| Ecallantide (Kalbitor) | Kallikrein inhibitor | 60-amino acid recombinant protein, blocks plasma kallikrein | SC | 30 mg SC | Risk of anaphylaxis (3%); requires medical supervision |
| Fresh Frozen Plasma (FFP) | Contains C1-INH | Replaces C1-INH by providing plasma proteins | IV | 2 units | Used when specific therapies unavailable; theoretical risk of worsening (contains kallikrein substrates) |
Epinephrine, antihistamines, corticosteroids - NOT effective (do NOT delay definitive treatment by using these)
10b. Short-term Prophylaxis (Before procedures: dental, endoscopy, surgery, intubation)
- C1-INH concentrate (most preferred, fastest acting)
- Danazol (attenuated androgen) - 2.5 mg/kg/day beginning 5-10 days before
- Tranexamic acid - antifibrinolytic; inhibits plasminogen → reduces C1 activation (see biochemistry diagram)
10c. Long-term Prophylaxis (Prevention of recurrent attacks)
| Drug | Class | MOA | Route | Dosing | Notes |
|---|---|---|---|---|---|
| C1-INH concentrate (Cinryze, Haegarda) | Replacement | Restores C1-INH levels | IV or SC | Every 3-4 days | SC self-administration available |
| Lanadelumab (Takhzyro) | Monoclonal antibody | Anti-plasma kallikrein antibody; blocks kallikrein activity | SC | 300 mg every 2 weeks | Most effective prophylaxis agent |
| Berotralstat (Orladeyo) | Oral kallikrein inhibitor | Small molecule; blocks plasma kallikrein | PO | 150 mg daily | First oral agent for HAE prophylaxis |
| Danazol (attenuated androgen) | Androgen | ↑ hepatic C1-INH synthesis from the normal allele | PO | Lowest effective dose | ADRs: virilization, hepatotoxicity, polycythemia; avoid in children/pregnancy |
| Tranexamic acid / ε-aminocaproic acid | Antifibrinolytic | Inhibits plasminogen → reduces C1 activation | PO | - | Less effective than androgens; contraindicated in thrombotic tendency/atherosclerosis |
Type III HAE does NOT respond to C1-INH replacement but may respond to danazol (which increases functional C1-INH) or progestin-only contraceptives (avoid combined OCP as estrogen worsens it).
11. Drug Target Map - Tying Biochemistry to Pharmacology
C1-INH ← Replaced by: C1-INH concentrate (plasma-derived or recombinant)
← Upregulated by: Danazol, Stanozolol (attenuated androgens)
FXIIa ← Blocked by: Anti-FXIIa monoclonal antibodies (in development: garadacimab)
← Reduced substrate: Tranexamic acid (↓ plasmin → ↓ C1 activation → ↓ FXIIa)
Plasma Kallikrein ← Inhibited by:
- Ecallantide (recombinant kallikrein inhibitor, acute)
- Lanadelumab (anti-kallikrein MAb, prophylaxis)
- Berotralstat (oral kallikrein inhibitor, prophylaxis)
Bradykinin B2 receptor ← Blocked by:
- Icatibant (decapeptide B2 antagonist, acute)
Kininase II (= ACE) ← Inhibited by ACE inhibitors → ↑bradykinin [AVOID in HAE!]
12. Acquired C1-INH Deficiency (Acquired Angioedema, AAE)
Differs from HAE:
- Onset after 4th decade (no family history)
- Clinically identical to HAE
- No pruritus/urticaria
Two types:
| Type I (AAE-I) | Type II (AAE-II) | |
|---|---|---|
| Cause | C1q activation by lymphoproliferative disorders, plasma cell dyscrasias → C1-INH consumed | Auto-antibodies against C1-INH |
| Associated disease | B-cell lymphoma, myeloma, Waldenström, SLE | Autoimmune |
| C1q | ↓ | ↓ |
| C4 | ↓ | ↓ |
Treatment: Address underlying malignancy/autoimmune disease; same acute/prophylaxis agents as HAE.
13. ACE Inhibitor-Induced Angioedema (Bradykinin-mediated)
Mechanism:
- ACE = Kininase II - normally degrades bradykinin
- ACE inhibitors block kininase II → bradykinin accumulates → B2 receptor activation → angioedema
- Incidence: ~0.1-0.7% of ACE inhibitor users; higher in Black patients and the elderly
- Usually orofacial - tongue and lip swelling predominant
- Can be life-threatening (laryngeal edema)
- Occurs anytime during treatment, even after years of tolerating the drug
Treatment:
- Stop ACE inhibitor immediately
- Icatibant or C1-INH concentrate (evidence is mixed but used in severe cases)
- Switch to ARB (angiotensin receptor blocker) if antihypertensive needed - ARBs do NOT affect bradykinin metabolism
Exam pearl: C4 is NORMAL in ACE inhibitor-induced angioedema (unlike HAE where C4 is always low)
14. Summary Comparison - Angioedema Types
| Feature | Allergic/Histamine-mediated | HAE (Type I/II) | ACE inhibitor-induced |
|---|---|---|---|
| Mediator | Histamine, leukotrienes | Bradykinin | Bradykinin |
| Urticaria | YES | NO | NO |
| Pruritus | YES | NO | NO |
| C4 | Normal | Always LOW | Normal |
| C1-INH | Normal | LOW/dysfunctional | Normal |
| Response to antihistamines | YES | NO | NO |
| Response to epinephrine | YES | NO (but used in emergency) | NO |
| Treatment | Antihistamines, steroids, epinephrine | C1-INH, icatibant, ecallantide | Stop ACE inhibitor, icatibant |
| GI involvement | Rare | Common | Less common |
| Laryngeal involvement | Possible | HIGH RISK | Possible (orofacial predominant) |
Sources:
- Dermatology 2-Volume Set 5e (Boliviana), Ch. 18 - p.376, Figs. 18.6
- Andrews' Diseases of the Skin, Ch. on HAE
- Harrison's Principles of Internal Medicine 22e (2025), Ch. 363
- Katzung's Basic and Clinical Pharmacology 16e, Ch. 17 (Kallikrein-Kinin system)
- Fitzpatrick's Dermatology 9e, Ch. 41
- Quick Compendium of Clinical Pathology 5e, §6.3.8.1
- Janeway's Immunobiology 10e
This is a shared conversation. Sign in to Orris to start your own chat.