Explain acute lymphoblastic leukemia from Robbins pathology textbook for 2nd year MBBS
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the content needed from Robbins. Let me compile the comprehensive answer.
Acute Lymphoblastic Leukemia/Lymphoma (ALL)
Robbins, Cotran & Kumar - Pathologic Basis of Disease
Definition
Acute lymphoblastic leukemia/lymphomas (ALLs) are neoplasms composed of immature B (pre-B) or T (pre-T) cells, called lymphoblasts. The term "leukemia" is used when the bone marrow and blood are heavily involved, while "lymphoma" is used when a mass lesion is the dominant presentation (e.g., a thymic mass).
- ~85% are B-ALLs - typically childhood acute leukemias
- ~15% are T-ALLs - tend to present in adolescent males as thymic lymphomas
Epidemiology
- ALL is the most common cancer of children
- ~2,500 new cases/year in the United States
- Peak incidence of B-ALL: ~3 years of age (correlates with the peak number of normal bone marrow pre-B cells)
- Peak of T-ALL: adolescence (when the thymus reaches maximum size)
- Slightly more frequent in boys than girls
- Hispanic/Latino children have the highest incidence of any ethnic group in the US
- Both B-ALL and T-ALL also occur (less frequently) in adults
Fig. 13.5 - Origin of Lymphoid Neoplasms
This diagram shows where B-ALL and T-ALL arise in the lymphoid differentiation hierarchy:
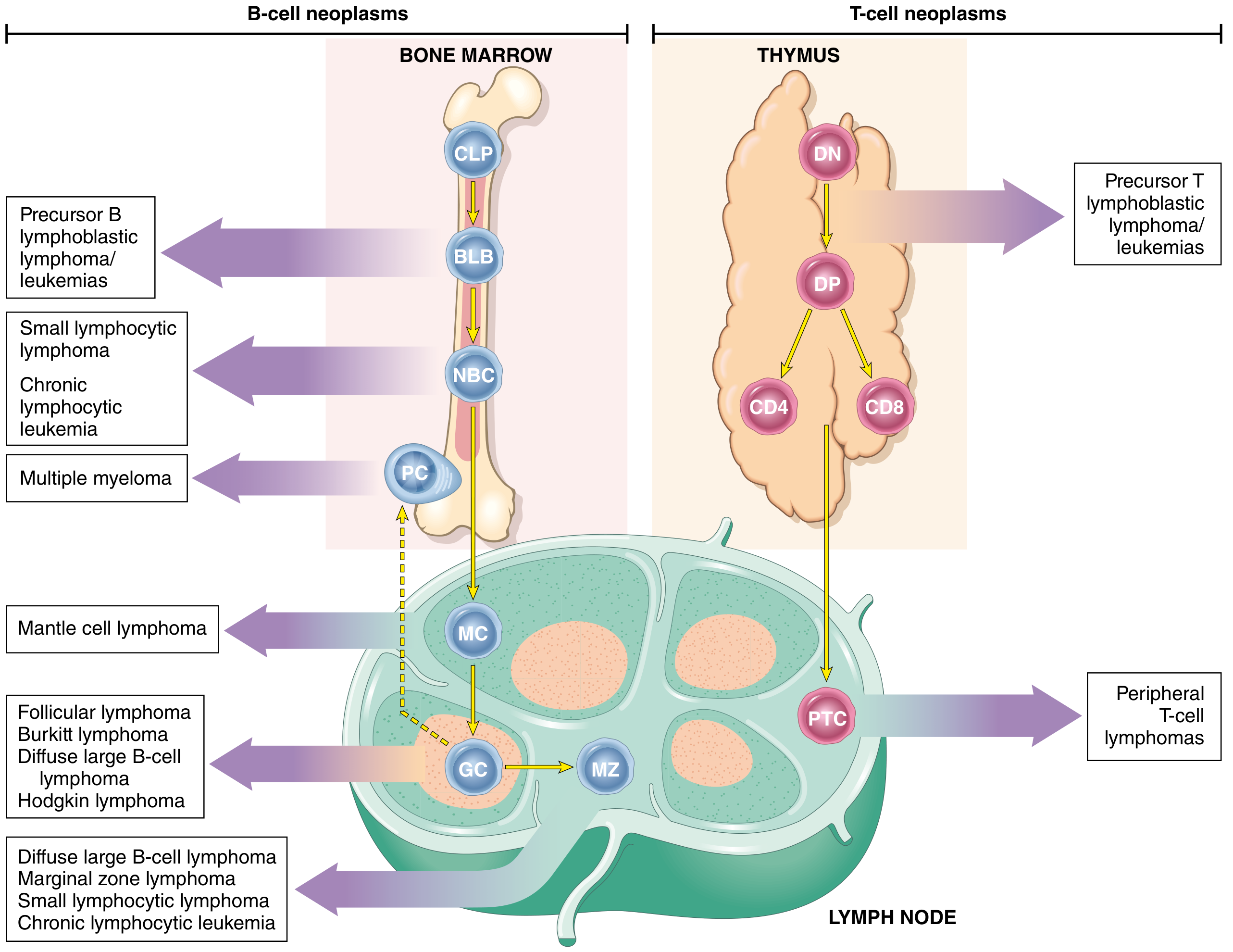
B-ALL arises from BLB (pre-B lymphoblasts) in the bone marrow. T-ALL arises from DN (double-negative) or DP (double-positive) pre-T cells in the thymus.
Pathogenesis
Transcription Factor Mutations (Core Mechanism)
Many chromosomal aberrations in ALL dysregulate transcription factors required for normal B- and T-cell development:
| ALL Type | Key Mutated Genes |
|---|---|
| T-ALL | NOTCH1 (50-70%) - essential for T-cell development |
| B-ALL | PAX5, TCF3, ETV6, RUNX1, BCR::ABL1, KMT2A, PBX1 |
These mutations cause maturation arrest (cells cannot differentiate) and increased self-renewal - a stem cell-like phenotype. The result is accumulation of immature, non-functional blasts.
Multistep Origin
Transcription factor mutations alone are not sufficient - ALL requires complementary driver mutations that promote cell growth:
- Mutations increasing tyrosine kinase activity (e.g., BCR-ABL)
- RAS signaling mutations
- Deep sequencing suggests fewer than 10 mutations are sufficient to produce full-blown ALL
Key Chromosomal Aberrations (~90% of ALLs have detectable abnormalities)
| Aberration | Gene Fusion | Significance |
|---|---|---|
| t(12;21) | ETV6::RUNX1 | Present in 25% of B-ALL; favorable prognosis |
| t(9;22) - Philadelphia chromosome | BCR::ABL tyrosine kinase | Present in B-ALL; BCR-ABL is 190 kDa (stronger kinase than 210 kDa form in CML); targeted by kinase inhibitors |
| Translocations involving KMT2A | KMT2A (MLL) fusions | Associated with infant ALL; poor prognosis |
| Hyperdiploidy (>50 chromosomes) | Trisomies of chr 4, 7, 10 | Favorable prognosis |
| NOTCH1 mutations (T-ALL) | - | Present in 50-70% of T-ALL |
Morphology
Gross / Bone Marrow
- Hypercellular marrow packed with lymphoblasts, replacing normal marrow elements
- Mediastinal (thymic) masses in 50-70% of T-ALL
- T-ALL more likely to have lymphadenopathy and splenomegaly than B-ALL
Microscopy (Fig. 13.6A)
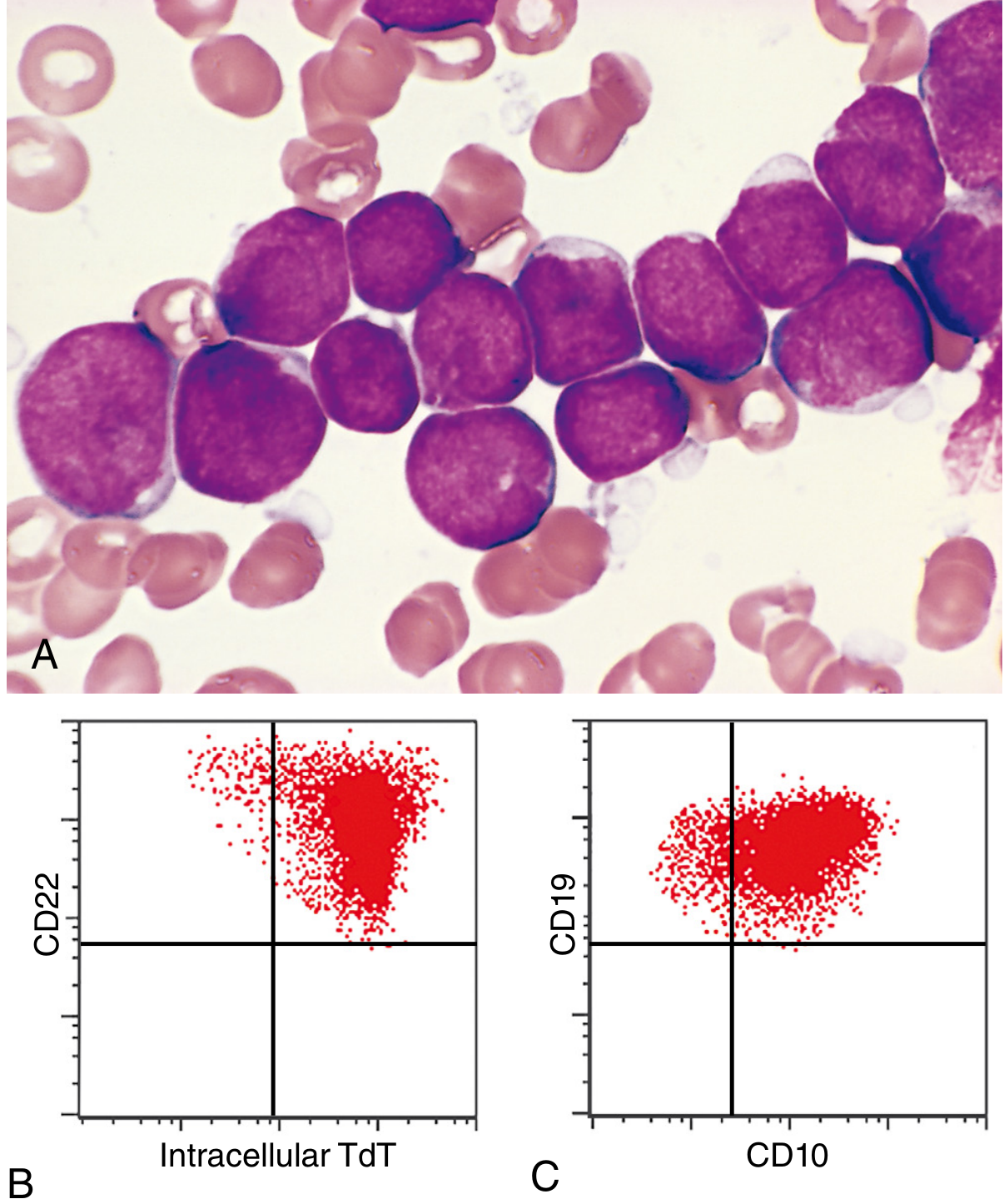
Fig. 13.6: (A) Lymphoblasts with condensed nuclear chromatin, small nucleoli, and scant agranular cytoplasm. (B) TdT+/CD22+ by flow cytometry. (C) CD10+/CD19+ confirming B-ALL.
Key histological features of lymphoblasts:
- Scant basophilic cytoplasm
- Nuclei slightly larger than small lymphocytes
- Delicate, finely stippled nuclear chromatin
- Small, sharply demarcated nucleoli
- Convoluted (deeply subdivided) nuclear membrane
- High mitotic rate (reflects aggressive behavior)
- Interspersed macrophages ingesting apoptotic cells may create a "starry sky" appearance
ALL vs. AML (Distinguishing Features)
| Feature | Lymphoblasts (ALL) | Myeloblasts (AML) |
|---|---|---|
| Chromatin | More condensed | Less condensed |
| Nucleoli | Less conspicuous | More prominent |
| Cytoplasm | Less, no granules | More, may have granules (Auer rods) |
| Myeloperoxidase | Negative | Positive |
| PAS stain | Positive (cytoplasmic material) | Variable |
Immunophenotype
TdT (Terminal Deoxynucleotidyl Transferase)
- A specialized DNA polymerase expressed only in pre-B and pre-T lymphoblasts
- Positive in >95% of ALL - hallmark marker
- Negative in mature lymphoid neoplasms and AML
B-ALL Markers
- CD19 (pan-B marker) + PAX5 transcription factor
- CD10 (CALLA - common ALL antigen) - present in most B-ALLs
- More mature B-ALLs also express CD20 and cytoplasmic IgM (μ heavy chain)
- Very immature B-ALLs may be CD10 negative
T-ALL Markers
- CD1, CD2, CD5, CD7
- Immature T-ALLs: negative for surface CD3, CD4, CD8
- "Late" pre-T cell tumors: positive for CD3, CD4, and CD8
Clinical Features
Onset is abrupt and stormy - symptoms develop within days to a few weeks:
1. Bone Marrow Failure (Pancytopenia)
- Fatigue (anemia - decreased RBCs)
- Fever and infections (neutropenia - decreased WBCs)
- Bleeding (thrombocytopenia - decreased platelets)
2. Mass Effects from Neoplastic Infiltration (more common in ALL than AML)
- Bone pain - from marrow expansion and subperiosteal infiltration
- Generalized lymphadenopathy
- Splenomegaly and hepatomegaly
- Testicular enlargement
- In T-ALL: complications from mediastinal mass (SVC syndrome, airway/vascular compression)
3. CNS Manifestations (more common in ALL)
- Headache, vomiting, nerve palsies from meningeal spread
- CNS is a "sanctuary site" - requires specific CNS-directed therapy (intrathecal chemotherapy)
Prognosis and Treatment
Treatment of pediatric ALL is one of oncology's great success stories:
- ~95% of children achieve complete remission with chemotherapy
- 75-85% are cured
- Adults have worse outcomes - less than half are cured
Poor Prognostic Factors
| Factor | Reason |
|---|---|
| Age < 2 years (infantile ALL) | Strong association with KMT2A translocations and CNS involvement |
| Adolescence or adulthood | Different molecular pathogenesis; adults cannot tolerate intensive chemo |
| Blast count >100,000/mm³ | High tumor burden |
Favorable Prognostic Factors
- Age 2 to 10 years
- Low white cell count
- Hyperdiploidy (>50 chromosomes)
- Trisomy of chromosomes 4, 7, and 10
- t(12;21) - ETV6::RUNX1 fusion gene
Special Considerations
- Minimal residual disease (MRD): Molecular detection of residual disease after therapy predicts worse outcome and guides treatment decisions
- Philadelphia chromosome-positive B-ALL [t(9;22)]: BCR-ABL kinase inhibitors (imatinib, dasatinib) + conventional chemotherapy - highly effective
- CAR-T cell therapy: Chimeric antigen receptor T cells directed against CD19 have produced dramatic responses in B-ALL; associated with significant toxicity and high cost
KEY CONCEPTS (Robbins Summary Box)
- Most common cancer in children; may be derived from precursor B or T cells. B-ALL = 75-80% of cases.
- Highly aggressive tumors manifesting with signs of bone marrow failure or rapidly growing masses.
- Tumor cells contain genetic lesions that block differentiation, leading to accumulation of immature, non-functional blasts.
- A subset contains activating mutations in tyrosine kinases (e.g., BCR-ABL) that are important therapeutic targets.
Source: Robbins, Cotran & Kumar - Pathologic Basis of Disease, Chapter 13: Diseases of White Blood Cells, Lymph Nodes, Spleen, and Thymus, pp. 556-559
What is fab and who classification
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
I now have all the source material needed. Here is the complete, well-structured answer:
FAB and WHO Classification of ALL
Part 1 - FAB (French-American-British) Classification
The FAB classification was proposed in 1976 by a cooperative group of French, American, and British hematologists. It classifies ALL purely on morphological (cell appearance) criteria, dividing it into three subtypes: L1, L2, and L3.
Important note: The FAB classification of ALL no longer holds prognostic relevance in modern practice. It has been largely replaced by the WHO classification, which is based on immunophenotype and cytogenetics. However, FAB is still asked in examinations.
FAB L1 - Most Common in Children (~85% of childhood ALL)
| Feature | Description |
|---|---|
| Cell size | Small to intermediate - about twice the size of a normal lymphocyte |
| N:C ratio | Very high (nucleus dominates the cell) |
| Nuclear chromatin | Partially condensed, relatively uniform |
| Nucleoli | Indistinct or absent |
| Cytoplasm | Scant, slightly basophilic |
| Nuclear shape | Regular, round to oval |
- This is the "classic" lymphoblast morphology
- Best prognosis among FAB subtypes
- Most common in children aged 2-10 years
FAB L2 - Most Common in Adults
| Feature | Description |
|---|---|
| Cell size | Large, heterogeneous (variable) |
| N:C ratio | Lower than L1 |
| Nuclear chromatin | Irregular, variable |
| Nucleoli | Prominent, one or more |
| Cytoplasm | More abundant than L1 |
| Nuclear shape | Irregular, with clefting and indentations |
| Cytoplasmic vacuoles | May be present |
- More heterogeneous appearance - cells vary in size
- T-ALL often shows greater morphologic heterogeneity with nuclear irregularities (resembles L2)
- No morphologic feature truly distinguishes B-ALL from T-ALL lineage
FAB L3 - Burkitt-type ALL (Rare, ~1-3%)
| Feature | Description |
|---|---|
| Cell size | Large, uniform |
| N:C ratio | Moderate |
| Nuclear chromatin | Finely stippled ("punched out") |
| Nucleoli | Prominent, multiple |
| Cytoplasm | Deeply basophilic, prominent vacuoles |
| Nuclear shape | Round to oval, regular |
- Morphologically identical to Burkitt lymphoma cells
- Corresponds to mature B-cell (surface IgM positive) - actually a mature B-cell neoplasm, not a true precursor ALL
- Associated with c-MYC translocations [t(8;14), t(2;8), t(8;22)]
- Worst prognosis in old FAB system
- In the modern WHO classification, this is classified as Burkitt lymphoma, not ALL
FAB Summary Table
| Feature | L1 | L2 | L3 |
|---|---|---|---|
| Cell size | Small | Large, variable | Large, uniform |
| N:C ratio | Very high | Moderate-low | Moderate |
| Nucleoli | Absent/indistinct | Prominent | Very prominent |
| Cytoplasm | Scant | More | Deeply basophilic + vacuoles |
| Nuclear shape | Regular | Irregular/clefted | Regular |
| Common in | Children | Adults | Rare (Burkitt) |
| Prognosis (old) | Good | Intermediate | Poor |
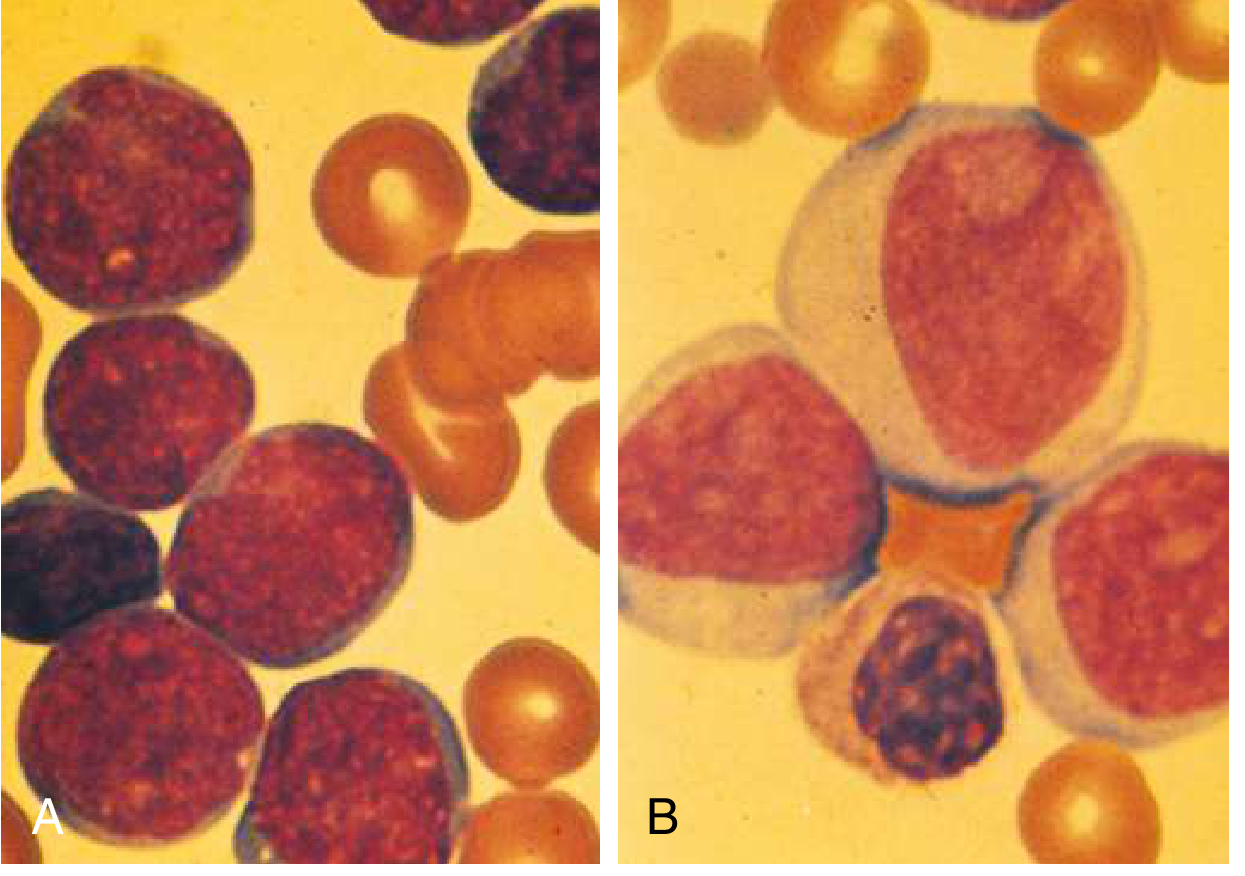
Comparison: ALL lymphoblasts (A) are smaller with higher nuclear-to-cytoplasmic ratio and less distinct nucleoli compared to AML myeloblasts (B) which show "punched out" clear nucleoli.
Part 2 - WHO Classification of ALL
The WHO classification is the modern, clinically used system. It integrates:
- Immunophenotype (flow cytometry markers)
- Cytogenetics (chromosomal translocations)
- Molecular genetics (fusion genes)
- Clinical features
This system is used because specific genetic subtypes carry distinct prognoses and different therapeutic targets (e.g., BCR-ABL can be targeted with imatinib).
A. B-Lymphoblastic Leukemia/Lymphoma (B-ALL)
1. B-ALL, NOS (Not Otherwise Specified)
- Diagnosed when none of the specific recurrent genetic abnormalities are found
- Still TdT+, CD19+, CD10+
2. B-ALL with Recurrent Genetic Abnormalities
| WHO Subtype | Translocation/Abnormality | Incidence | Prognosis |
|---|---|---|---|
| B-ALL with BCR-ABL1 | t(9;22)(q34.1;q11.2) - Philadelphia chromosome | 3-5% pediatric; 25% adult | Poor (but improved with TKIs) |
| B-ALL with KMT2A rearranged | t(v;11q23.3) - KMT2A (MLL) fusions | 5% pediatric | Poor (especially infant ALL) |
| B-ALL with ETV6-RUNX1 | t(12;21)(p13.2;q22.1) - cryptic, FISH needed | 25% pediatric, 3% adult | Very good |
| B-ALL with Hyperdiploidy | >50 chromosomes; trisomies of chr 4, 7, 10 | 25% pediatric | Very good |
| B-ALL with Hypodiploidy | 24-44 chromosomes | 1% | Variable (near-haploid = very poor) |
| B-ALL with IL3-IGH | t(5;14)(q31.1;q32.3) | 1% | Intermediate |
| B-ALL with TCF3-PBX1 | t(1;19)(q23;p13.3) | 5% | Intermediate |
| Provisional: B-ALL, BCR-ABL1-like | Gene expression like Ph+, but Philadelphia chromosome absent; JAK2, CRLF2 mutations | 10-20% pediatric; 20-30% adult | Poor |
| Provisional: B-ALL with iAMP21 | Intrachromosomal amplification of chromosome 21 | 2% | Poor |
B. T-Lymphoblastic Leukemia/Lymphoma (T-ALL)
- Presents predominantly in adolescent males
- Associated with mediastinal (thymic) mass in 50-70% of cases
- Markers: CD1, CD2, CD5, CD7; TdT+; variable CD3, CD4, CD8
| WHO Subtype | Feature |
|---|---|
| T-ALL, NOS | Main category; NOTCH1 mutations in 50-70% |
| Provisional: Early T-cell Precursor ALL (ETP-ALL) | Immature T-cell arrested at very early stage; lacks CD1a/CD8; high-risk subtype |
| Provisional: NK-cell lymphoblastic leukemia/lymphoma | Natural killer cell origin |
Why WHO is Superior to FAB
| Aspect | FAB | WHO |
|---|---|---|
| Basis | Morphology only | Immunophenotype + cytogenetics + genetics |
| Prognostic value | Limited/none | High - specific subtypes predict outcome |
| Therapeutic guidance | None | Identifies targetable mutations (BCR-ABL → TKI therapy) |
| Detection method | Light microscopy | Flow cytometry, FISH, PCR, karyotype |
| Current use | Exams / historical | Clinical practice standard |
Quick Exam Memory Aid
FAB = Fmorphology only → L1 (children, good), L2 (adults, medium), L3 (Burkitt, worst)
WHO = Whole picture (genes + markers) → Named by translocation (e.g., ETV6-RUNX1 = good prognosis; BCR-ABL = targeted therapy available)
Sources: Tietz Textbook of Laboratory Medicine, 7th Ed.; Goldman-Cecil Medicine; Robbins Pathologic Basis of Disease
This is a shared conversation. Sign in to Orris to start your own chat.