Neurocutaneous syndrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
neurocutaneous syndromes phakomatoses review
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Loading Image
Loading Image
Finding Sources
Reading File
Loading Image
I now have all the material needed for a thorough, well-illustrated answer. Here is the complete coverage:
Neurocutaneous Syndromes (Phakomatoses)
Neurocutaneous syndromes are congenital, often hereditary disorders that feature lesions of both the skin and the nervous system. Because both structures share a common ectodermal origin, the same genetic defect disrupts their development simultaneously. The term phakomatosis (from Greek phakos = "lens-shaped spot") is a synonym still used in neuroradiology and ophthalmology. The most clinically important members are NF-1, NF-2, tuberous sclerosis complex, Sturge-Weber syndrome, and von Hippel-Lindau disease.
- Goldman-Cecil Medicine, p. 4049
- Grainger & Allison's Diagnostic Radiology, p. 1985
1. Neurofibromatosis Type 1 (NF-1)
Genetics and Epidemiology
- Most common neurocutaneous syndrome; incidence 1 in 3,000-4,000 births
- Also the most common autosomal dominant condition overall
- Gene: NF1 on chromosome 17q11.2, encoding neurofibromin (a RAS-GAP tumor suppressor)
- 50% of cases represent new (de novo) mutations
Diagnostic Criteria (at least 2 of the following)
| Feature | Details |
|---|---|
| Cafe-au-lait macules | ≥6 macules (>5 mm prepubertal; >15 mm postpubertal) |
| Neurofibromas | ≥2 cutaneous/subcutaneous, or 1 plexiform |
| Axillary/inguinal freckling | Crowe sign |
| Optic pathway glioma | WHO grade I pilocytic astrocytoma (up to 15% of patients) |
| Lisch nodules | Iris hamartomas (pathognomonic) |
| Sphenoid wing dysplasia | Orbital/skeletal dysplasia |
| Affected first-degree relative |
CNS Manifestations
- Optic pathway gliomas (OPGs): most common brain lesion; more often affect optic nerves than the chiasm in NF-1. Most are asymptomatic; chiasmatic involvement risks precocious puberty and visual loss.
- Plexiform neurofibromas (pathognomonic when large), peripheral nerve gliomas
- Learning disability, attention deficit disorder, cerebral vasculopathy
Other Organ Involvement
- Pheochromocytoma, renal artery stenosis, macrocephaly, skeletal abnormalities (pseudoarthrosis of tibia)
- Risk of malignant peripheral nerve sheath tumors (MPNSTs)
Imaging
MRI: Fusiform expansion of the optic nerve, widening of the optic foramen; T2 "UBOs" (unidentified bright objects) in basal ganglia and cerebellum (do not enhance, not true tumors).
Treatment
-
Mirdametinib (MEK inhibitor) - FDA-approved 2025 for plexiform neurofibromas in NF-1 (PMID 40419758)
-
Selumetinib (MEK inhibitor) also used for inoperable plexiform neurofibromas
-
Annual ophthalmologic exam; blood pressure monitoring; developmental screening
-
Grainger & Allison's Diagnostic Radiology, pp. 1985-1987
-
Goldman-Cecil Medicine, p. 4050
2. Neurofibromatosis Type 2 (NF-2)
Genetics
- Gene: NF2 on chromosome 22q12, encoding merlin (schwannomin), a tumor suppressor
- Autosomal dominant; less common than NF-1
Hallmark Features
- Bilateral vestibular schwannomas (cranial nerve VIII) - pathognomonic; cause progressive bilateral sensorineural deafness
- Multiple meningiomas, ependymomas, astrocytomas
- Plaque-like intracutaneous tumors; cafe-au-lait macules (~40%)
- Posterior subcapsular cataracts (lens opacities in young adults)
Treatment
-
Surgical resection of schwannomas (hearing preservation difficult even with early surgery)
-
Bevacizumab (anti-VEGF, 5 mg/kg IV every 2 weeks) can improve hearing in some patients
-
Goldman-Cecil Medicine, pp. 4051-4052
3. Tuberous Sclerosis Complex (TSC)
Genetics
- Incidence ~1 in 6,000; autosomal dominant; up to 75% are sporadic mutations
- TSC1 gene at 9q34.3 → encodes hamartin
- TSC2 gene at 16p13.3 → encodes tuberin
- Both proteins together inhibit mTOR (mammalian target of rapamycin); mutations cause mTOR upregulation → uncontrolled cell growth, neuronal migration defects, angiogenesis
- TSC2 mutations tend to produce more severe phenotype
Multi-organ Manifestations
| Organ | Lesion |
|---|---|
| Brain | Cortical tubers, subependymal nodules, subependymal giant cell astrocytoma (SEGA) |
| Skin | Facial angiofibromas, "ash leaf" hypomelanotic macules, shagreen patch, ungual fibromas |
| Kidney | Angiomyolipomas |
| Heart | Cardiac rhabdomyomas (present neonatally, may regress) |
| Lung | Lymphangioleiomyomatosis (LAM; predominantly in women) |
| Eye | Retinal hamartomas |
Diagnostic Criteria
Definite TSC: 2 major features, or 1 major + 2 minor features.
Major features: facial angiofibromas/forehead plaque, ungual fibromas, >3 hypomelanotic macules, shagreen patch, multiple retinal hamartomas, cortical tuber, subependymal nodule, SEGA, cardiac rhabdomyoma, LAM, renal angiomyolipoma.
Minor features: dental enamel pits, hamartomatous rectal polyps, bone cysts, cerebral white matter radial migration lines, gingival fibromas, "confetti" skin lesions.
MRI of Tuberous Sclerosis
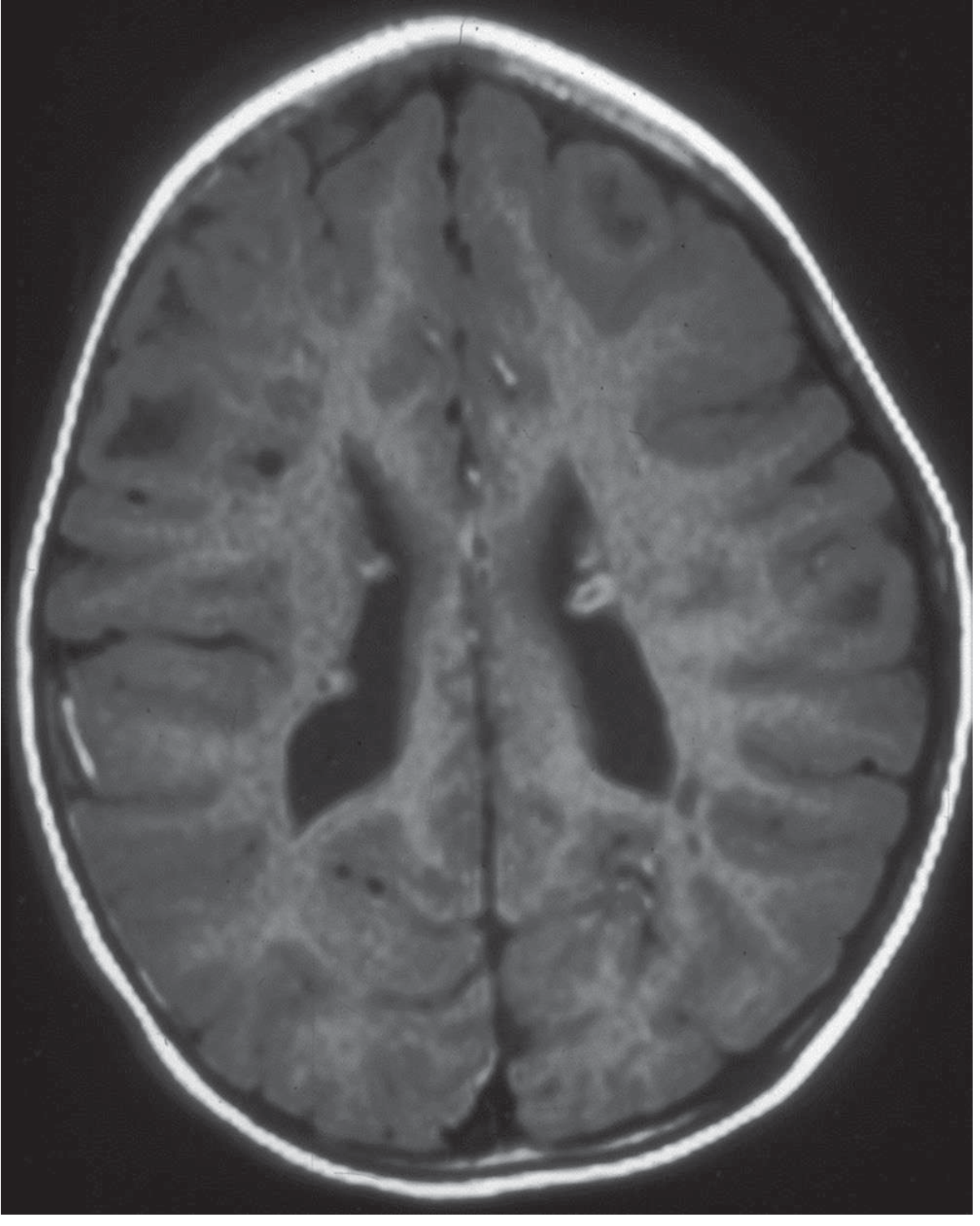
Treatment
-
mTOR inhibitors have dramatically altered prognosis:
- Everolimus (10 mg/day): reduces angiomyolipoma size in ~40%; reduces SEGA by ≥50% in ~1/3 of patients
- Everolimus also approved for intractable epilepsy due to TSC
- Lesions regrow when medication is stopped
- Topical rapamycin (0.1%) shrinks facial angiofibromas
- Sirolimus (2 mg/day) approved for LAM
-
Vigabatrin is first-line for infantile spasms in TSC
-
Surgical resection if seizure focus localized to cortical dysplasia
-
Goldman-Cecil Medicine, pp. 4049-4050
4. Sturge-Weber Syndrome (Encephalotrigeminal Angiomatosis)
Genetics
- Sporadic (not familial); incidence ~1 in 20,000
- Caused by a somatic mosaic mutation in GNAQ (chromosome 9q21) - not detectable on blood-based genetic testing
Classic Triad
- Port-wine stain (capillary hemangioma) of upper face in the distribution of cranial nerve V1 (ophthalmic branch)
- Ipsilateral leptomeningeal angioma (usually parieto-occipital)
- Ocular abnormalities: glaucoma, choroidal hemangioma
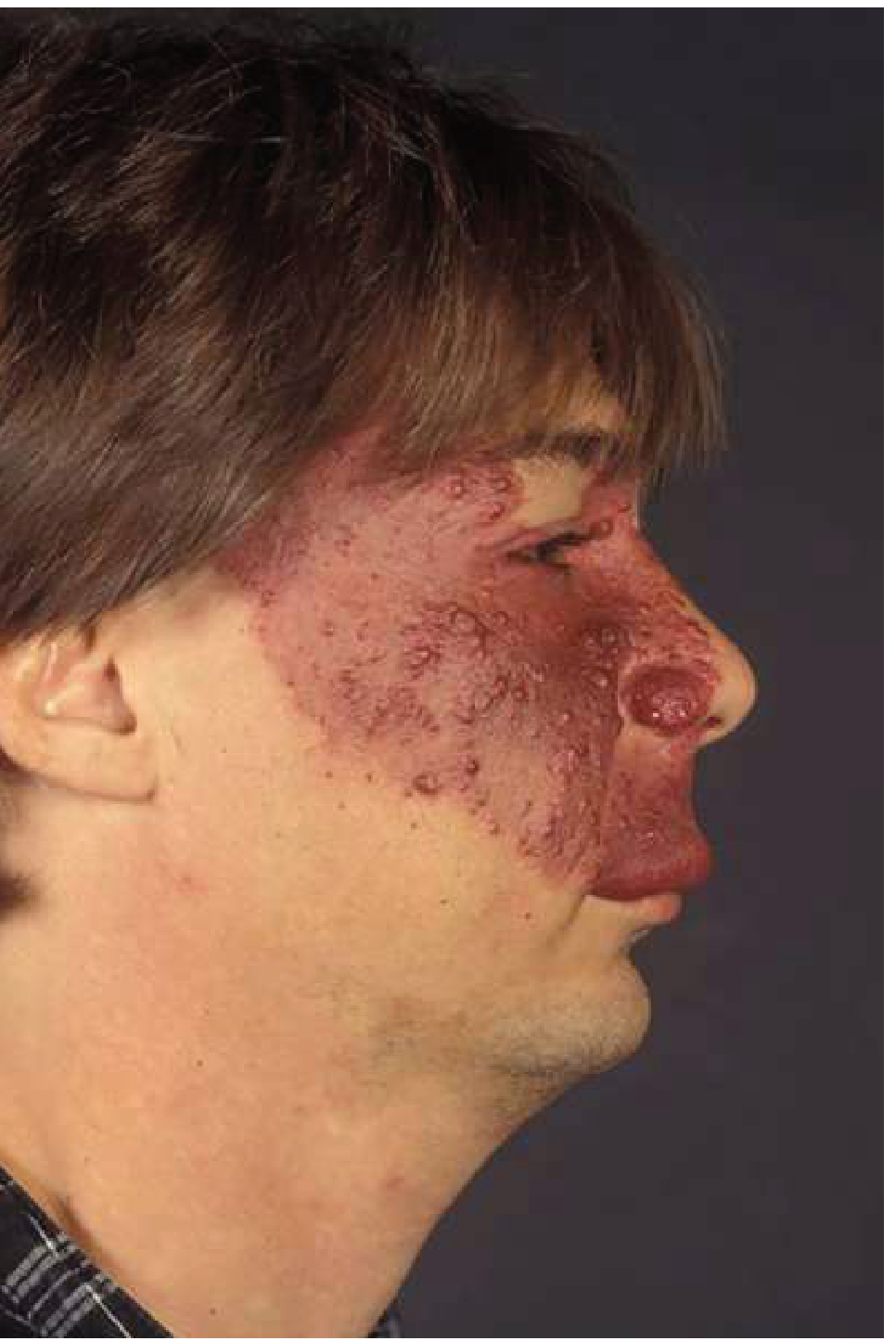
Neurologic Features
- Focal-onset epilepsy (most common neurologic manifestation)
- Variable cognitive impairment, hemiparesis, hemianopia
- Classic "tramline" (double-contour) cortical calcification on CT (may be absent in infants)
- Only individuals with port-wine stain of the upper face and periorbital area develop leptomeningeal angioma; only ~15% of people with an upper facial nevus have brain involvement
MRI Findings
- Leptomeningeal enhancement (pial angioma) on contrast T1
- Cortical atrophy of the affected hemisphere
- Enlarged ipsilateral choroid plexus
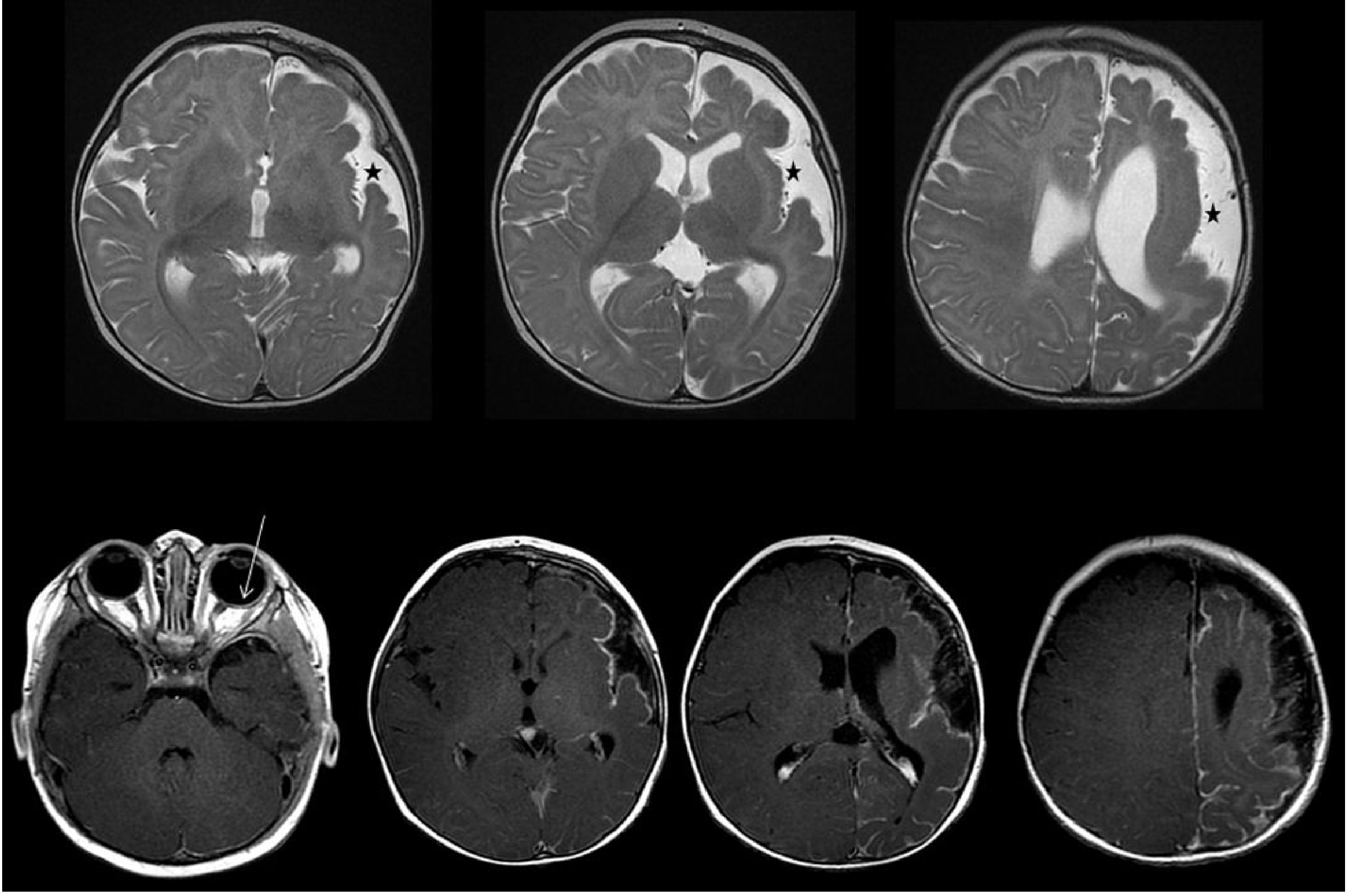
Treatment
-
Laser therapy for port-wine stain
-
Antiepileptic drugs; surgical excision of epileptogenic cortex if refractory
-
Periodic glaucoma screening throughout life (glaucoma can occur even without neurologic involvement)
-
Goldman-Cecil Medicine, pp. 4052-4053
-
Kanski's Clinical Ophthalmology, p. 2263
5. Von Hippel-Lindau (VHL) Disease
Genetics
- Autosomal dominant; gene: VHL tumor suppressor at chromosome 3p25-26
- VHL protein normally targets HIF (hypoxia-inducible factor) for degradation; loss of VHL leads to HIF overactivation and VEGF overproduction → angiogenesis
Clinical Features (onset typically 3rd-4th decade)
| System | Lesion |
|---|---|
| CNS | Hemangioblastomas of cerebellum, brainstem, spinal cord |
| Eye | Retinal angiomas (can cause retinal detachment) |
| Kidney | Renal cell carcinoma (clear cell type), bilateral renal cysts |
| Adrenal | Pheochromocytoma |
| Pancreas | Pancreatic cysts, neuroendocrine tumors |
| Ear | Endolymphatic sac tumors |
| Liver/Epididymis | Cysts |
VHL Subtypes
| Subtype | Features |
|---|---|
| Type 1 | CNS/retinal hemangioblastoma + RCC; no pheochromocytoma |
| Type 2A | Hemangioblastoma + pheochromocytoma; low RCC risk |
| Type 2B | All three (hemangioblastoma + pheo + RCC) |
| Type 2C | Pheochromocytoma only |
Diagnosis
- More than one brain hemangioblastoma, OR one hemangioblastoma + visceral manifestation, OR one manifestation + positive family history
- Genetic testing detects VHL mutation in essentially all affected individuals
Surveillance & Treatment
-
Annual ophthalmologic exam, annual BP monitoring, pheochromocytoma screening every 5 years, abdominal ultrasound from age 16 years
-
Surgical resection of brain tumors, RCC, pheochromocytoma; gamma knife for smaller CNS tumors
-
Pazopanib (tyrosine kinase inhibitor, 800 mg/day) for progressive lesions
-
Goldman-Cecil Medicine, pp. 4053-4054
6. Ataxia-Telangiectasia (Louis-Bar Syndrome)
Genetics
- Autosomal recessive; gene: ATM (ataxia telangiectasia mutated) at 11q22.3
- ATM encodes a serine/threonine kinase activated by DNA double-strand breaks; loss leads to defective DNA repair
Clinical Features
-
Progressive cerebellar ataxia (onset in early childhood, Purkinje cell degeneration)
-
Oculocutaneous telangiectasias (conjunctivae, ears, nasal mucosa, antecubital/popliteal fossae)
-
Recurrent sinopulmonary infections (combined T + B cell immunodeficiency; deficient IgA)
-
Oculomotor apraxia
-
High risk of malignancies (leukemia, lymphoma, breast cancer in ATM heterozygote females)
-
Radiosensitivity (avoid X-ray radiation)
-
Elevated serum AFP and CEA; MRI shows cerebellar atrophy
-
Quick Compendium of Clinical Pathology, p. 3462
-
Bradley and Daroff's Neurology in Clinical Practice
7. Summary Table of Key Neurocutaneous Syndromes
| Syndrome | Inheritance | Gene/Chromosome | Key Skin Feature | Key Neurologic Feature |
|---|---|---|---|---|
| NF-1 | AD | NF1/17q11.2 | Cafe-au-lait macules, cutaneous neurofibromas, axillary freckling | Optic glioma, Lisch nodules, plexiform neurofibromas |
| NF-2 | AD | NF2/22q12 | Plaque-like tumors, cafe-au-lait (~40%) | Bilateral vestibular schwannomas |
| Tuberous sclerosis | AD | TSC1/9q34 or TSC2/16p13 | Ash leaf macules, angiofibromas, shagreen patch, ungual fibromas | Cortical tubers, SEGA, infantile spasms |
| Sturge-Weber | Sporadic (somatic) | GNAQ/9q21 | Port-wine stain (V1 distribution) | Leptomeningeal angioma, epilepsy, glaucoma |
| Von Hippel-Lindau | AD | VHL/3p25-26 | None specific | Cerebellar hemangioblastoma, retinal angioma |
| Ataxia-telangiectasia | AR | ATM/11q22.3 | Oculocutaneous telangiectasias | Progressive cerebellar ataxia |
Key Points to Remember
- The phakomatoses share a common ectodermal origin of skin and nervous system - hence both are affected
- NF-1 is diagnosed clinically (cafe-au-lait + neurofibromas + Lisch nodules); MEK inhibitors (mirdametinib/selumetinib) are now first-line for plexiform neurofibromas
- TSC management is transformed by mTOR inhibitors (everolimus, sirolimus)
- Sturge-Weber is caused by a somatic (non-heritable) GNAQ mutation - only 15% of port-wine stains involve the brain
- VHL = think hemangioblastoma + RCC + pheochromocytoma; VEGF pathway is the therapeutic target
- Ataxia-telangiectasia: the only AR member in this group; always screen for malignancy and avoid ionizing radiation
Recent update (2025): Mirdametinib received FDA approval for NF-1-related plexiform neurofibromas (Hoy SM, Drugs 2025; PMID 40419758).
PMLE
Reading File
Finding Sources
Finding Sources
Searching PubMed
polymorphic light eruption
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Reading File
I now have comprehensive content across three major dermatology/medicine textbooks. Let me compile the complete answer.
Polymorphic Light Eruption (PMLE)
Synonyms: Polymorphous light eruption, benign summer light eruption, prurigo aestivalis (historical)
PMLE is the most common photodermatosis, accounting for the majority of photosensitivity complaints seen in clinical practice. It is an immunologically mediated, delayed-type hypersensitivity (DTH) reaction to UV-induced skin antigens in susceptible individuals.
Epidemiology
-
Prevalence 10-20% of the general population in temperate climates
-
Prevalence is inversely related to latitude: highest in Scandinavia (~22%), high in the UK and northern US (10-15%), low in Australia (~5%), and rare near the equator (~1% in Singapore) - explained by year-round UV exposure producing sustained immunologic tolerance ("hardening") in sunny climates
-
Female predominance (~4:1); onset typically in the 2nd and 3rd decades
-
Occurs in all skin types and all racial groups; pinhead papular variant linked to darker Fitzpatrick types (African American, Asian)
-
Skin type I carries highest risk; skin type IV the lowest in pan-European studies
-
Up to 70% of the population may carry a genetic susceptibility, though not all express clinical disease due to variable penetrance
-
Fitzpatrick's Dermatology, p. 1641
-
Dermatology 2-Volume Set 5e, p. 1865
Pathogenesis
The central mechanism is a resistance to UV-induced immunosuppression, leading to a persistent DTH response against photoinduced endogenous cutaneous antigens.
Normal UV Response (absent in PMLE)
In healthy skin, UV radiation causes:
- Depletion of epidermal Langerhans cells
- Neutrophilic infiltration into skin
- Release of immunosuppressive cytokines (IL-4, IL-10) - a Th2 micromilieu
- Net effect: suppression of contact hypersensitivity and induction of hapten-specific tolerance
What Goes Wrong in PMLE
- Langerhans cells persist (CD1a+ cells are not depleted after UV exposure)
- Neutrophil infiltration is impaired - chemotaxis is defective
- Reduced IL-4 and IL-10 production
- Shift to a Th1 cytokine profile rather than the normal Th2 profile
- This creates a permissive environment for hypersensitivity against UV-modified skin molecules
- Elevated baseline levels of IL-1 family pro-inflammatory cytokines are found in PMLE
Photoantigens
The specific UV-induced antigen(s) have not been identified. UV irradiation modifies proteins and DNA, generating novel antigens. UV-irradiated epidermal cells from PMLE patients stimulate autologous peripheral blood mononuclear cells more strongly than cells from healthy controls - indirect evidence of photoantigen formation.
Why "Hardening" Works
Photohardening therapy (repeated low-dose UV exposure) restores the impaired Langerhans cell depletion and neutrophil influx, re-establishing the normal immunosuppressive Th2 milieu.
- Fitzpatrick's Dermatology, pp. 1645-1646
- Dermatology 5e, p. 1865
Clinical Features
Trigger & Timing
- Triggered by UVA, UVB, and rarely visible light - from sunlight, tanning beds, or phototherapy units
- Action spectrum: predominantly UVA (most common), UVB, rarely visible light
- Lesions appear within minutes to hours (up to 2 days) after first significant UV exposure of the season
- Most severe in spring and early summer in temperate climates; tends to improve as summer progresses ("hardening")
- Resolves spontaneously within days to a week without scarring
Lesion Morphology
The name "polymorphic" (polymorphous) reflects wide inter-individual variation - each individual tends to develop the same morphology consistently ("monomorphic" within the same patient):
| Morphology | Features |
|---|---|
| Papular (most common) | Small erythematous papules; coalesce into plaques |
| Papulovesicular | Papules + small vesicles |
| Vesicular | Frank vesicles (less common) |
| Plaque type | Urticarial plaques; confluent involvement |
| Pinpoint papular | Pinhead-sized papules; common in darker phototypes |
Clinical Photo - Papular/Plaque Type
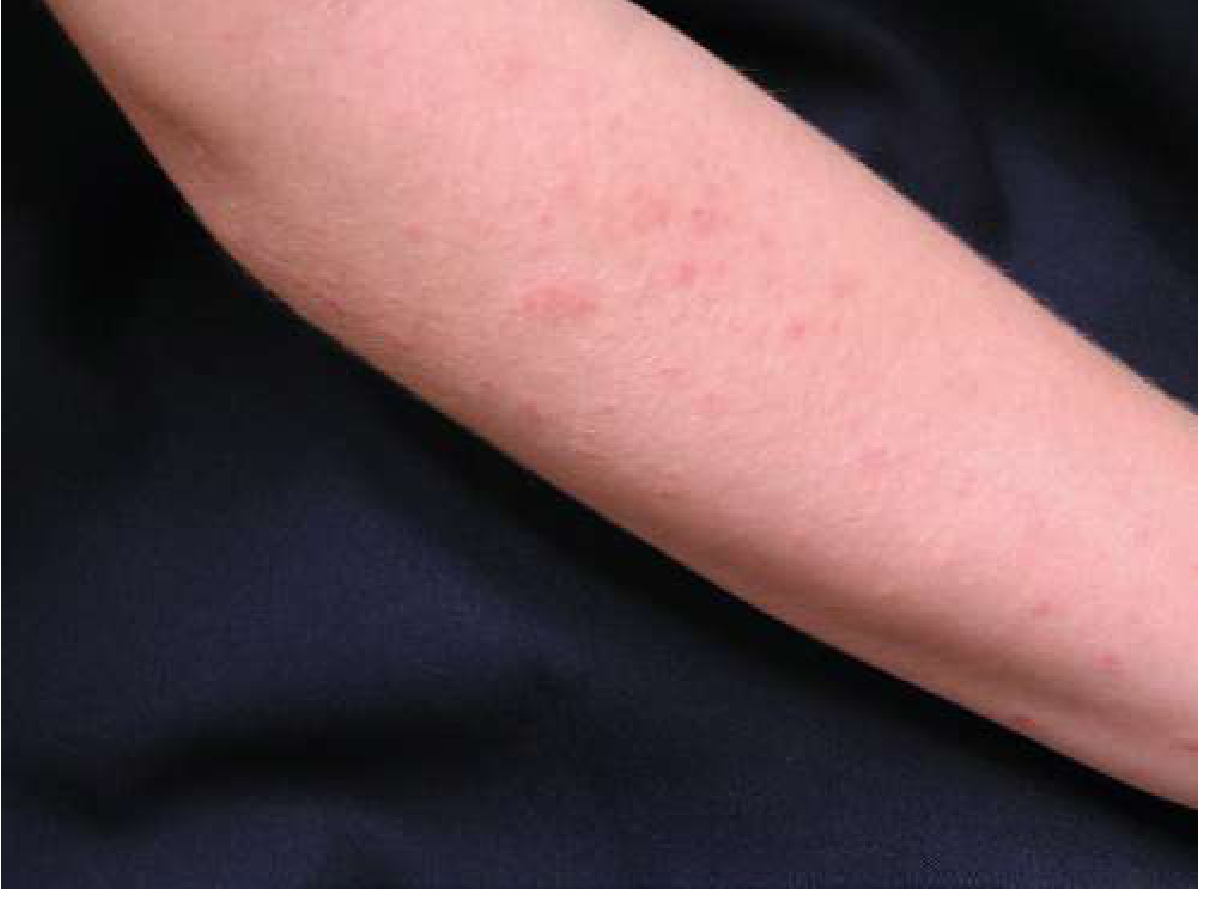
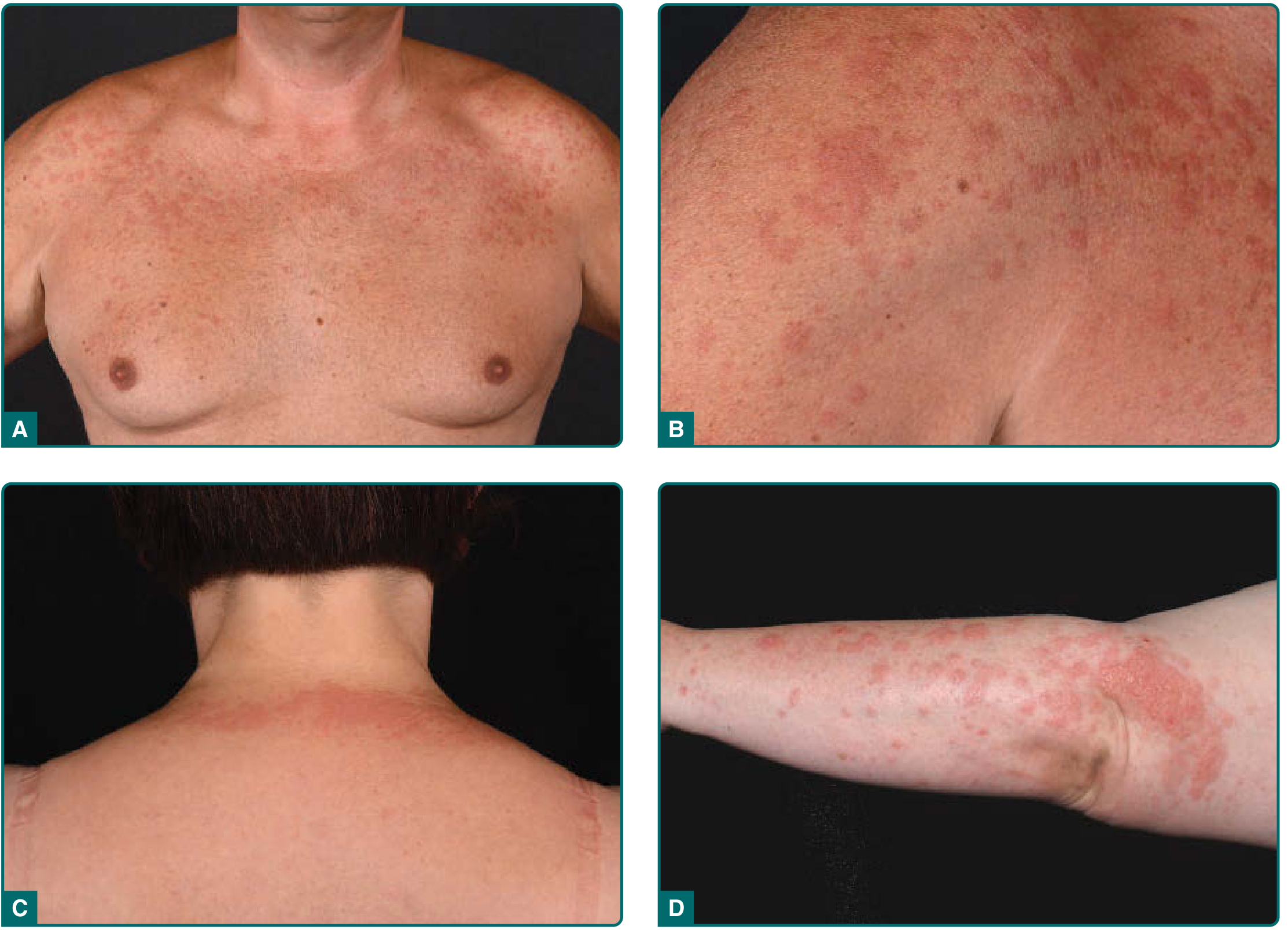
Distribution
- Symmetrically distributed on sun-exposed skin: dorsa of hands, forearms, chest, shoulders, V-neck area, lower legs
- Face is sometimes involved but may be relatively spared due to chronic daily UV exposure providing natural hardening
- Sun-protected areas are typically uninvolved
Symptoms
- Pruritus is a consistent feature - often intense
- No systemic symptoms (fever, malaise)
Histopathology
-
Epidermal spongiosis (intercellular edema)
-
Papillary dermal edema (prominent)
-
Superficial and deep perivascular mixed-cell infiltrate (lymphocytes, histiocytes)
-
Timed biopsy after solar-simulated irradiation: CD4+ T cells predominate early (within hours), followed by CD8+ T cells in established lesions
-
No epidermal necrosis, no interface change (helps distinguish from lupus)
-
Fitzpatrick's Dermatology, p. 4376
Clinical Variants
| Variant | Key Features |
|---|---|
| Juvenile spring eruption | Affects ears of young boys; occurs in spring; considered a PMLE variant |
| Pinpoint papular variant | Pinhead papules; predominant in dark-skinned individuals |
| Actinic prurigo | Severe persistent form; childhood onset; cheilitis + conjunctivitis; HLA-DR4 (DRB1*0407) association; common in Native Americans |
| Mallorca acne (acne aestivalis) | Acneiform follicular papules on neck/shoulders; linked to oily sunscreen vehicles |
Diagnosis
Clinical (primary approach)
- Characteristic history (spring onset, UV-triggered, resolves spontaneously, recurs annually) + typical morphology is sufficient for diagnosis in most cases
- No specific laboratory test
Investigations in Atypical Cases
- Phototesting: typically normal MED to UVB and UVA (unlike chronic actinic dermatitis); presence of PMLE is not reflected in abnormal MED
- Photoprovocation test: Repeated suberythemal doses of UVA or UVB reproduce PMLE lesions in 60-90% of affected patients; most sensitive to UVA
- Skin biopsy: helpful if diagnosis is uncertain; biopsy provoked lesions for highest yield
- ANA, anti-dsDNA, anti-Ro/La: to exclude lupus erythematosus (most important differential - especially subacute cutaneous LE)
Differential Diagnosis
| Condition | Distinguishing Feature |
|---|---|
| Lupus erythematosus (subacute cutaneous, discoid) | Positive ANA/ENA; interface dermatitis on biopsy; lesions persist 10-14 days after photoprovocation; photoprovocation negative area must be watched for weeks |
| Chronic actinic dermatitis | Older men; lichenified plaques; persistent; abnormal MED to UVB/UVA |
| Solar urticaria | Urticaria within minutes; resolves within 1-2 hours; triggered by visible light too |
| Actinic prurigo | Childhood onset; persistent year-round; cheilitis/conjunctivitis; HLA-DR4 |
| Hydroa vacciniforme | Childhood; papulovesicles/bullae; vacciniform scarring; EBV-associated |
| Phototoxicity/photoallergy | Drug/chemical exposure history; photopatch test positive |
| Lymphocytic infiltration (Jessner-Kanof) | Persistent for months-years; predominantly facial/auricular; no seasonal pattern |
Course and Prognosis
-
Chronic, episodic condition with slow tendency toward improvement over years
-
In a 32-year follow-up study: 24% of patients achieved complete resolution, 51% reported symptomatic improvement, 24% had equal or worsening symptoms
-
PMLE does not increase risk of skin cancer (though UV-induced immunosuppression in normal skin does)
-
"Hardening" during the season naturally reduces severity as summer progresses
-
Goldman-Cecil Medicine, p. 4053
-
Fitzpatrick's Dermatology, p. 4651
Management
1. Photoprotection (Mild Disease)
- Broad-spectrum sunscreen covering both UVA and UVB (SPF ≥30; high UVA protection is essential - moderate UVA protection is often insufficient)
- SPF 45 with high UVA protection applied at 1 mg/cm² can prevent UV-provoked PMLE in most patients
- Protective clothing, sun avoidance (especially during peak hours)
2. Acute Treatment
- Topical corticosteroids: reduce inflammation and itch; shorten eruption
- Oral antihistamines: adjunct for pruritus relief
- Short course of oral prednisolone (20-30 mg/day tapered over several days): effective for infrequent, severe flares (e.g., vacations); taken at first sign of pruritus
3. Prophylactic Photohardening (Moderate-Severe Disease)
The most effective preventive treatment for patients with frequent or severe PMLE:
- Narrowband UVB (NB-UVB): 3 sessions/week for 15-20 treatments in early spring; restores Langerhans cell depletion and neutrophil influx
- PUVA (psoralen + UVA): alternative to NB-UVB; similar efficacy
- Mechanism: re-establishes normal UV-induced immunosuppressive milieu in skin
4. Other Agents
- Hydroxychloroquine: evidence from controlled trial; useful in cases not responding to photohardening
- Afamelanotide (alpha-MSH analog): clinical trial evidence for prevention
- Oral antioxidants (nicotinamide, beta-carotene, Polypodium leucotomos extract): modest protective effect as supplements; a 2025 systematic review (PMID 39804624) found evidence for Polypodium leucotomos and other oral photoprotectants
- Biologics/JAK inhibitors: under investigation; a 2026 systematic review (PMID 41192435) evaluated monoclonal antibodies and JAK inhibitors in idiopathic photosensitive disorders - still emerging data
Key Points Summary
| Feature | Detail |
|---|---|
| Most common photodermatosis | 10-20% prevalence in temperate populations |
| Core mechanism | Resistance to UV-induced immunosuppression → DTH to photoantigens |
| Action spectrum | UVA > UVB; rarely visible light |
| Timing | Hours to days after first spring UV exposure; resolves in days |
| Morphology | Papular (most common); polymorphic between patients, monomorphic within |
| Key differential | Lupus erythematosus (must always exclude) |
| MED | Usually normal (distinguishes from chronic actinic dermatitis) |
| "Hardening" | Gradual improvement through season; basis of phototherapy treatment |
| First-line severe Rx | NB-UVB photohardening in early spring |
| Prognosis | Chronic; slow natural improvement; ~24% remit over 32 years |
Recent evidence: A 2025 immunopathogenesis review (PMID 41655098) highlights the role of effector/regulatory T cell imbalance and cytokine dysregulation (IL-1 family, Th1 predominance) as therapeutic targets - JAK inhibitors and biologics targeting this pathway are now under active investigation.
- Fitzpatrick's Dermatology Vol 1-2, Ch. 92
- Dermatology 2-Volume Set 5e, Ch. 87
- Goldman-Cecil Medicine, p. 4053
PMLE in retroviral therapy
Finding Sources
Finding Sources
Searching PubMed
polymorphic light eruption antiretroviral HIV
Finding Sources
Finding Sources
Searching PubMed
photosensitivity antiretroviral therapy HIV skin
PMLE caused by JC virus
Finding Sources
Searching PubMed
progressive multifocal leukoencephalopathy JC virus treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading Article
PMID: 37487750
Reading Article
PMID: 40621097
I have all the material needed. Let me now compile the full answer. First, an important clarification:
Important clarification: "PML" (Progressive Multifocal Leukoencephalopathy) and "PMLE" (Polymorphic Light Eruption) are two entirely different conditions. Your previous questions were about PMLE (a photodermatosis). The JC virus causes PML - a demyelinating brain disease. The similarity in abbreviations can create confusion. This answer covers PML caused by JC virus in full.
PML - Progressive Multifocal Leukoencephalopathy
Definition
PML is a demyelinating disease of the central nervous system caused by reactivation of the latent JC polyomavirus (John Cunningham virus) in immunocompromised individuals. The virus preferentially infects oligodendrocytes, so demyelination is its principal pathologic effect.
- Bradley and Daroff's Neurology in Clinical Practice, p. 1640
- Robbins Pathologic Basis of Disease, p. 1170
The JC Virus
- Named after John Cunningham, the patient from whom it was first isolated
- A small non-enveloped double-stranded DNA polyomavirus (Polyomaviridae family)
- Seroprevalence: 70-90% of adults have antibodies, indicating ubiquitous primary infection in childhood or early adulthood - usually completely asymptomatic
- Less than 10% of healthy individuals show evidence of ongoing viral replication
- JC virus establishes latency in the kidneys, bone marrow, lymphoid tissue, and possibly the brain
- PML is the only known clinical complication of JC virus infection
Epidemiology and Predisposing Conditions
PML occurs almost exclusively in immunocompromised patients. Classic settings include:
| Context | Details |
|---|---|
| HIV/AIDS | Most common cause historically; occurs in ~4-5% of AIDS patients; typically with CD4 <100/mm³ |
| Hematologic malignancies | Hodgkin's lymphoma, chronic lymphocytic leukemia (B-cell malignancies - first described context) |
| Monoclonal antibody therapy | Natalizumab (anti-α4-integrin, used in MS and Crohn's disease) - major modern cause |
| Other immunosuppressives | Rituximab, mycophenolate, tacrolimus |
| Organ transplantation | Immunosuppressive regimens |
| Granulomatous diseases | Sarcoidosis |
The incidence in HIV/AIDS has declined somewhat since the introduction of combination ART (cART), though less dramatically than other opportunistic infections. Natalizumab-associated PML became a major concern from 2005 onwards.
Pathogenesis
- Primary infection in childhood: asymptomatic; virus establishes latency in kidneys, bone marrow, lymphoid tissues
- Reactivation under immunosuppression: impaired JCV-specific CD4+ and CD8+ T cell surveillance allows viral replication
- Hematogenous spread to the CNS (mechanism debated - may involve infected B cells crossing the blood-brain barrier; natalizumab blocks lymphocyte trafficking via α4-integrin, possibly trapping JCV-infected cells in the CNS)
- Oligodendrocyte infection: JCV enters oligodendrocytes (and to a lesser extent astrocytes and cerebellar granule cells); viral replication causes lytic cell death
- Demyelination: loss of oligodendrocytes strips axons of myelin, producing expanding foci of demyelination
In natalizumab-associated PML, the integrin-blocking mechanism prevents JCV-specific CD4+ T cells from entering the brain, creating a CNS-specific immunodeficient state even in the setting of normal systemic immunity.
Clinical Features
- Onset: subacute to gradual; relentlessly progressive over weeks to months
- Symptoms depend on lesion location - predominantly subcortical white matter (parieto-occipital most common):
| Symptom | Frequency |
|---|---|
| Visual field defects (homonymous hemianopia) | Common (parieto-occipital predilection) |
| Hemiparesis | Common |
| Cognitive impairment / personality change | Common |
| Aphasia and language disorders | Moderate |
| Ataxia | Cerebellar involvement |
| Sensory deficits | Variable |
| Seizures | Occur; cortical involvement |
- No fever (distinguishes from bacterial/fungal infections)
- In HIV/AIDS PML: typically presents as a late AIDS complication
- In natalizumab PML: may present in a patient with otherwise normal immune status; often more inflammatory ("inflammatory PML")
Pathology
Gross
- Irregular, ill-defined white matter lesions ranging from millimeters to large confluent regions
- Predominantly subcortical distribution; spares the cortical ribbon ("scalloping out" of the grey-white border)
- Parieto-occipital lobes most commonly affected; cerebellum and brainstem also involved; spinal cord very rarely
Histology (Robbins classic triad)
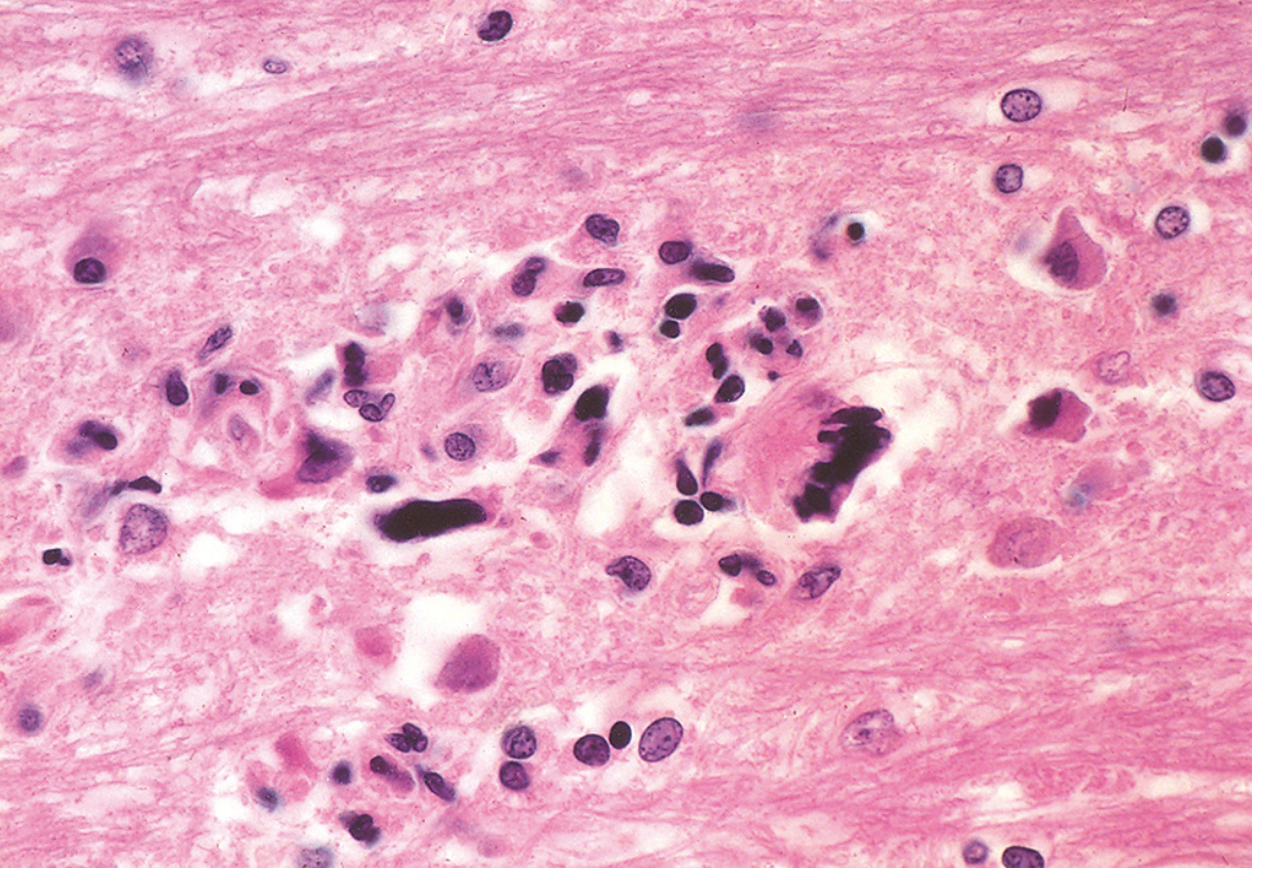
- Altered oligodendrocytes - greatly enlarged nuclei with glassy amphophilic intranuclear viral inclusions (found at lesion edge)
- Bizarre giant astrocytes - one to several irregular, hyperchromatic nuclei; mitotically active appearance
- Demyelination - lipid-laden macrophages in center of lesions; reduced axons
- Robbins Pathologic Basis of Disease, p. 1170
Neuroimaging
MRI (modality of choice)
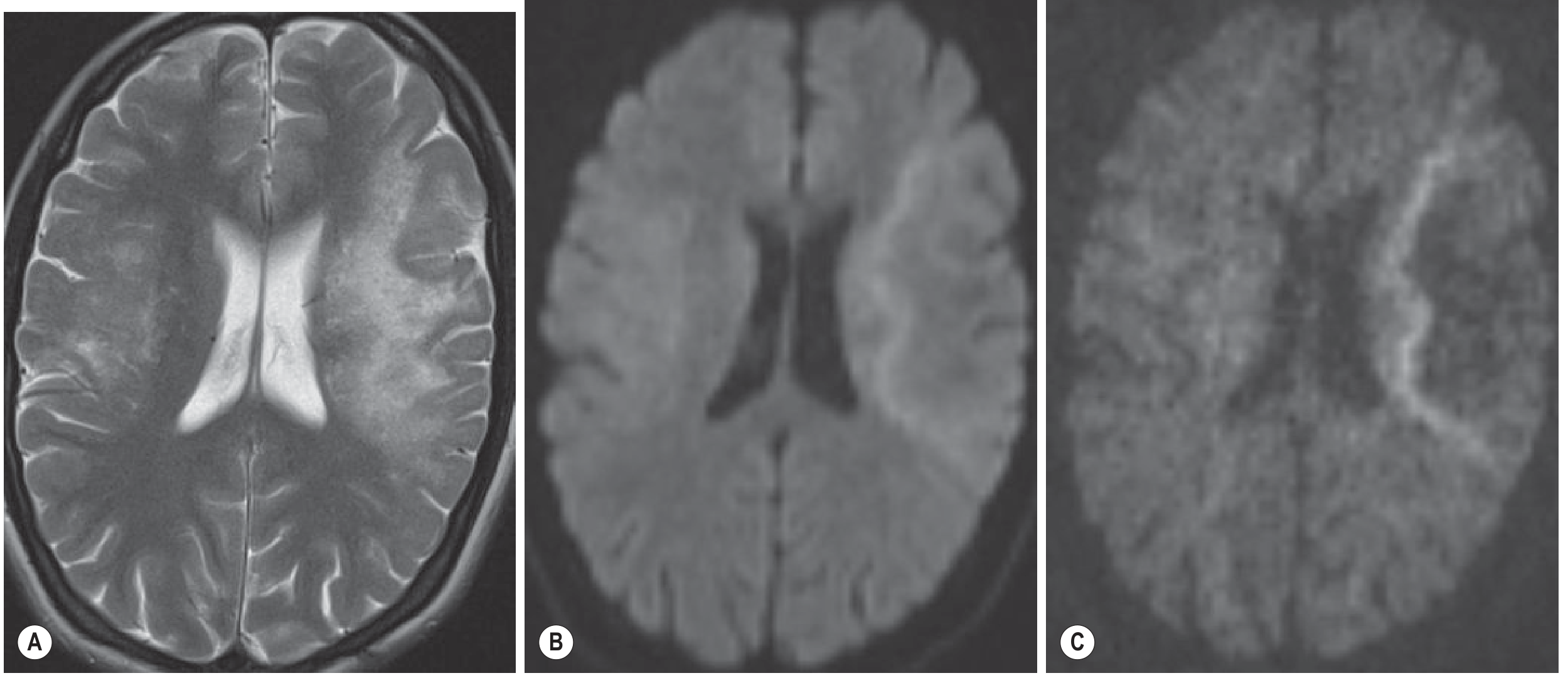
| Sequence | Finding |
|---|---|
| T2/FLAIR | Hyperintense white matter lesions; ill-defined borders; subcortical/periventricular; may be multifocal |
| T1 | Markedly hypointense (reflects severe tissue destruction) |
| DWI | Characteristic peripheral hyperintense rim at the advancing edge of demyelination (active zone); central area of completed necrosis is dark |
| Contrast (Gd) | Usually no enhancement (classic PML); however enhancement occurs in ~15% of HIV/AIDS cases and more frequently in immune reconstitution (IRIS) and natalizumab-PML |
| CT | Multifocal hypodense lesions with swelling; poor sensitivity vs. MRI |
- Grainger & Allison's Diagnostic Radiology, p. 1480
- Key pattern: Subcortical lesion that "scallops out" the grey-white border, sparing the cortex; no mass effect in classical PML
Diagnosis
CSF Analysis
- Usually normal or nonspecific (mild pleocytosis, mildly elevated protein)
- JC virus DNA PCR in CSF: highly specific; sensitivity ~70-90%. When positive in the correct clinical and MRI context, brain biopsy is not required.
- Negative CSF PCR does not exclude PML (especially post-treatment or inflammatory PML)
Brain Biopsy
- Required when CSF PCR is negative but clinical/imaging suspicion remains high
- Shows the histologic triad above; JCV immunohistochemistry or in situ hybridization confirms diagnosis
Natalizumab risk stratification - JCV antibody index
- Antibody index <0.4: considered negative (very low risk)
- 0.4-0.9: low risk
- 0.9-1.5: medium risk
- >1.5: high risk - consider drug switch
- Prior natalizumab exposure duration and prior immunosuppressant use multiply the risk
Treatment and Management
1. Restore Immune Competence (cornerstone)
HIV/AIDS:
- Immediate initiation or optimization of ART (combination antiretroviral therapy) is the most effective intervention
- ART restores CD4+ T cell count and JCV-specific T cell responses
- 1-year survival with ART: ~50% (pre-ART era: mean survival 2-4 months)
- Better outcomes with: CD4 >300/μL, low/undetectable HIV viral load, undetectable JCV in CSF after ART, contrast-enhancing lesions at diagnosis
Natalizumab-associated PML:
- Immediately discontinue natalizumab
- Accelerate drug elimination with plasma exchange (PLEX) - 5 sessions - to rapidly remove natalizumab and restore CNS immune surveillance
- Monitor closely for IRIS after drug removal
2. No Proven Specific Antiviral Therapy
Multiple agents have been tried in RCTs - none showed significant benefit:
- Cytarabine (cytosine arabinoside) - intravenous and intrathecal - failed RCT in HIV-PML
- Cidofovir - failed RCT in HIV-PML
- Mefloquine - prospective multicenter trial showed no benefit
- Mirtazapine (5-HT2A receptor antagonist, inhibits JCV binding to oligodendrocyte serotonin receptors) - case reports only; no RCT evidence
- Interferon-alpha - retrospective non-controlled data only
3. Immunotherapy (Emerging)
- Pembrolizumab (PD-1 inhibitor): small case series showed clinical improvement and stabilization by restoring anti-JCV T cell responses - promising but unvalidated
- JCV-specific cytotoxic T lymphocyte (CTL) infusions: positive results in small case series; currently experimental
- Both approaches target the fundamental problem: restoring JCV-specific cellular immunity
4. Managing IRIS (Immune Reconstitution Inflammatory Syndrome)
When ART (or natalizumab removal) restores immunity, a paradoxical inflammatory response to JCV antigens can occur:
- PML-IRIS: new or worsening CNS inflammation despite viral suppression
- Imaging: new contrast enhancement, increased FLAIR signal, mass effect, restricted diffusion - on a background of previously non-enhancing lesions
- CSF: pleocytosis
- Treatment: IV glucocorticoids (no controlled trial evidence; used empirically)
- PML-IRIS is actually associated with better long-term outcome than non-inflammatory PML - it reflects immune recovery
Prognosis
| Condition | Median Survival |
|---|---|
| Pre-cART HIV/AIDS | 2-4 months |
| HIV/AIDS + cART | ~24 months (50% survive 1 year) |
| Natalizumab-PML (drug stopped promptly) | Better than HIV-PML; up to 80% survive, but many with significant disability |
- Up to 80% of survivors have significant neurologic sequelae
- Up to 50% of long-term HIV/AIDS survivors (>24 months) experience meaningful recovery
- Favorable prognostic factors: higher CD4, low viral load, undetectable CSF JCV after treatment, presence of contrast enhancement (reflects intact immune response)
Summary
| Feature | PML |
|---|---|
| Cause | JC polyomavirus reactivation |
| Key cell infected | Oligodendrocytes |
| Effect | Demyelination |
| Classic population | AIDS (CD4 <100), natalizumab-treated MS patients |
| MRI hallmark | T2 hyperintense subcortical WM lesions; DWI peripheral rim; no enhancement (classic) |
| CSF test | JCV PCR (70-90% sensitivity) |
| Histology | Oligodendrocyte intranuclear inclusions + bizarre giant astrocytes + demyelination |
| Treatment | Restore immune competence (ART / stop immunosuppressant) |
| No proven antiviral | Cidofovir, cytarabine, mefloquine all failed RCTs |
| Emerging therapy | Pembrolizumab, JCV-specific CTL infusions |
Recent evidence: A 2023 Neurology review (PMID 37487750) emphasizes that restoring JCV-specific cellular immunity is the critical determinant of viral clearance and survival, and calls for validated biomarkers and definitive clinical trials. A 2025 review on natalizumab-PML (PMID 40621097) outlines the current JCV antibody index-based risk stratification as the standard of care for risk mitigation in natalizumab-treated patients.
Pericardial effusion
Finding Sources
Searching PubMed
pericardial effusion diagnosis management guidelines
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Reading Article
PMID: 40767817
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Loading Image
I now have comprehensive content from five major textbooks with imaging. Here is the complete answer:
Pericardial Effusion
Definition and Normal Physiology
The pericardium consists of two serous layers enclosing a potential space. Normally it contains 15-35 mL of fluid (some sources cite 5-10 mL detectable by echo). Any volume >50 mL is considered abnormal. The pericardium's semirigid enclosure affects pressure distribution across cardiac chambers and mediates right-left ventricular interaction during diastolic filling - properties that become critically important when fluid accumulates.
- Grainger & Allison's Diagnostic Radiology, p. 359
- Textbook of Clinical Echocardiography, p. 306
Etiology
Pericardial effusion can result from virtually any process affecting the pericardium:
| Category | Examples |
|---|---|
| Idiopathic | Most common in acute pericarditis (~85% in developed countries) |
| Viral infection | Coxsackievirus B, Epstein-Barr, CMV, echovirus, HIV |
| Bacterial | Staphylococcus, Streptococcus pneumoniae, tuberculosis (especially immunocompromised) |
| Fungal/Parasitic | Echinococcus, Candida, Aspergillus |
| Neoplastic | Lung Ca (direct extension), breast Ca, lymphoma, melanoma - metastatic >>primary; ~20% of large unexplained effusions are undiagnosed cancer |
| Autoimmune / Inflammatory | Lupus, RA, scleroderma, Dressler syndrome (post-MI autoimmune), post-pericardiotomy |
| Uremia | Dialysis patients |
| Cardiac surgery / procedures | Iatrogenic; post-cardiac intervention |
| Radiation | Mediastinal/chest irradiation; can be immediate or delayed by years |
| Drug-induced | High-dose anthracyclines, cyclophosphamide, hydralazine, procainamide |
| Trauma | Blunt or penetrating; aortic dissection (retrograde hemorrhage) |
| Transudative | Congestive heart failure, renal failure, hepatic insufficiency, hypothyroidism |
| HIV | Both direct and from opportunistic infections; higher risk of tamponade |
Key point: Bacterial/fungal infection, HIV, and malignancy carry higher risk of progressing to tamponade.
- Mulholland & Greenfield's Surgery, p. 4572
- Textbook of Clinical Echocardiography, Table 10.1
Pathophysiology
The hemodynamic significance of a pericardial effusion depends on two factors:
- Volume of fluid
- Rate of accumulation
A slowly expanding effusion may grow to >1000 mL with minimal hemodynamic effect (gradual pericardial stretch accommodates the volume). Rapid accumulation of even 50-100 mL can cause marked increases in pericardial pressure and acute tamponade.
Pressure-Volume Relationship
When fluid accumulates rapidly, the steep portion of the pericardial compliance curve is reached quickly - small additional volumes cause large pressure rises. Slow accumulation allows time on the flat compliance curve.
Cardiac Tamponade Physiology
-
Tamponade occurs when pericardial pressure exceeds intracardiac chamber pressure, impairing filling
-
Low-pressure thin-walled chambers (atria) are compressed before ventricles
-
Right heart is affected first → underfilling of left heart → reduced cardiac output
-
Tamponade generally occurs when filling pressures reach 15-20 mm Hg
-
Can occur at lower pressures in hypovolemia (dialysis, diuretics, hemorrhage)
-
The body compensates via adrenergic response (tachycardia, increased contractility) - beta-blockers impair this compensation
-
Textbook of Clinical Echocardiography, p. 307
-
Mulholland & Greenfield's Surgery, p. 4572
Clinical Features
Uncomplicated Pericardial Effusion
- Often asymptomatic, discovered incidentally on imaging
- Symptoms when present: dyspnea (especially positional), chest heaviness, cough (from bronchial compression), dysphagia
- Large chronic effusions may be well-tolerated with few symptoms
Cardiac Tamponade - Clinical Signs
Beck's Triad (classic but each component may be absent):
- Hypotension (reduced cardiac output)
- Jugular venous distension (JVD) - elevated venous pressure
- Muffled heart sounds
Additional Signs:
- Tachycardia - sensitivity 100% for tamponade
- JVD - sensitivity 100% for tamponade
- Pulsus paradoxus >10 mmHg fall in systolic BP during inspiration
- Sensitivity 98%, specificity 83%; LR+ 5.9, LR- 0.03
- Mechanism: inspiratory RV expansion compresses LV in a fixed pericardial space, reducing LV stroke volume
- Absent in: LV dysfunction, ASD, positive-pressure ventilation, aortic regurgitation, regional tamponade
- Loss of y-descent on JVP waveform (tricuspid cannot open freely; blood only enters when blood leaves)
- Diaphoresis, anxiety
Differentials to consider: right heart failure, pulmonary embolism (overlapping presentations).
- Symptom to Diagnosis Guide, p. 5180
- Mulholland & Greenfield's Surgery, p. 4573
Investigations
1. ECG
- Sinus tachycardia - most common finding
- Low voltage - attenuation of complexes by surrounding fluid
- Diffuse ST elevation and PR depression (if associated pericarditis)
- Electrical alternans - pathognomonic for large effusion/tamponade; alternating QRS axis/amplitude due to the heart swinging back and forth within the effusion
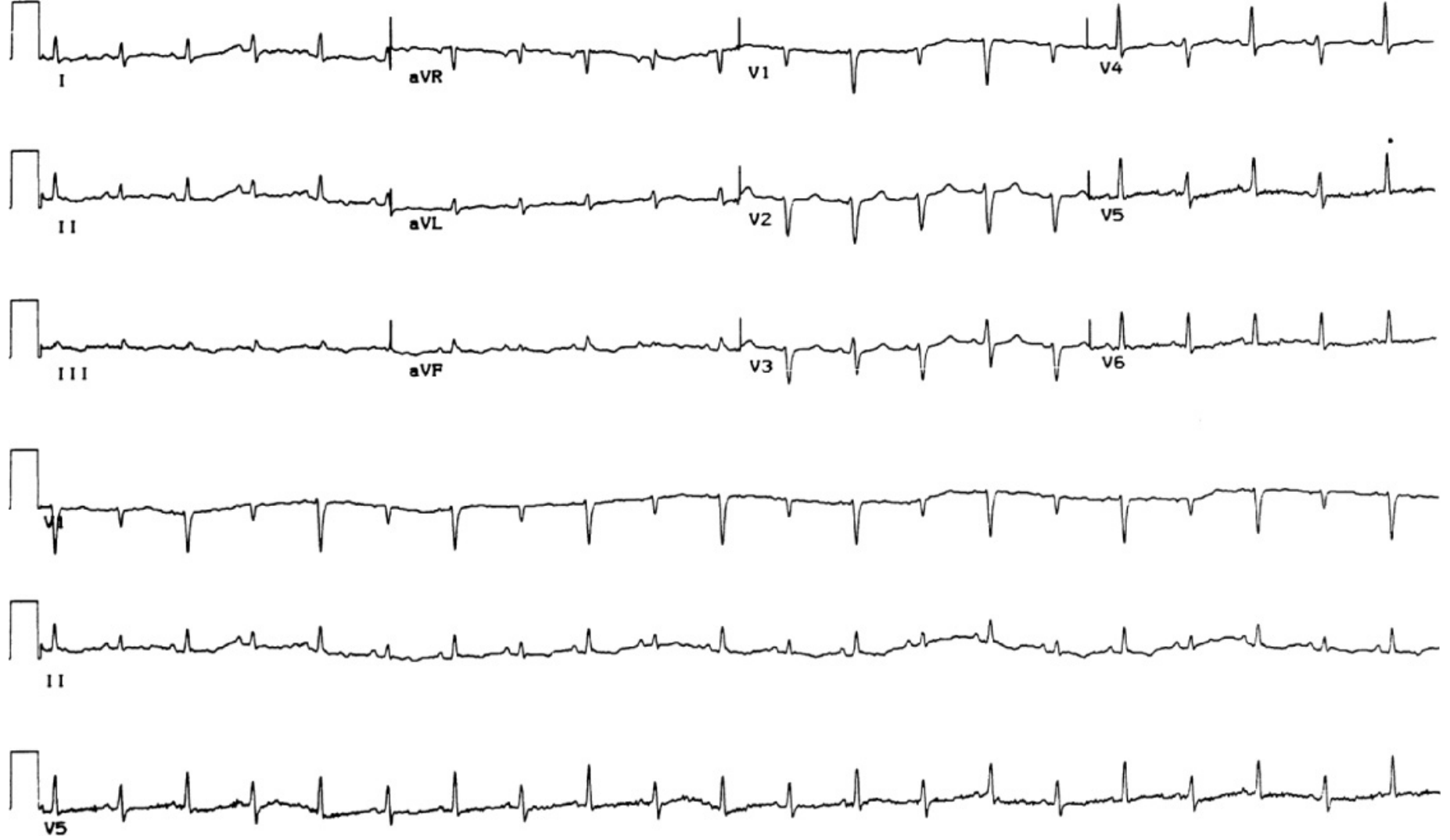
2. Chest X-Ray
- Normal when <200 mL of fluid
- "Water-bottle" or flask-shaped cardiac silhouette when ≥200 mL
- Symmetric enlargement with acute cardiophrenic angles
- Hilar vessels obscured (unlike simple cardiomegaly where hila are conspicuous)
- Posterior pericardial fat pad sign on lateral view: fat stripe displaced posteriorly
- Curvilinear lucency along the left cardiac border (pericardial fat line)
- Rapid change in heart size with no change in pulmonary vascular pattern (fluid accumulates faster than pulmonary congestion can develop)
3. Echocardiography (primary imaging investigation)
Echocardiography is the first-line and most useful test for pericardial effusion.
Detection:
- Echo-lucent (anechoic) space around the heart
- Small effusion: seen only posterior to LV free wall in systole
- Moderate-large effusion: circumferential, surrounds the heart
- Distribution is gravity-dependent (posterolateral LV wall, inferior to RV, superior pericardial recess)
- Important pitfall: Anterior epicardial fat can mimic a small effusion (fine speckled pattern rather than truly anechoic)
Size Classification (echo):
| Grade | Separation | Volume |
|---|---|---|
| Trivial/small | <10 mm posterior | <100 mL |
| Moderate | 10-20 mm | 100-500 mL |
| Large | >20 mm; anterior space >5 mm | >500 mL |
A distance >4 mm between pericardial leaflets is considered abnormal.
Echo Signs of Tamponade:
- Right atrial systolic collapse - earliest sign; sensitivity ~100%
- Right ventricular diastolic collapse - more specific; >1/3 diastole = significant
- IVC plethora - dilated IVC (<50% collapse with sniff)
- Exaggerated respiratory variation (Doppler): >25% decrease in mitral inflow velocity with inspiration (vs normal <10%); reciprocal increase in tricuspid inflow
- Ventricular interdependence: RV enlarges on inspiration while LV shrinks (septal bounce)
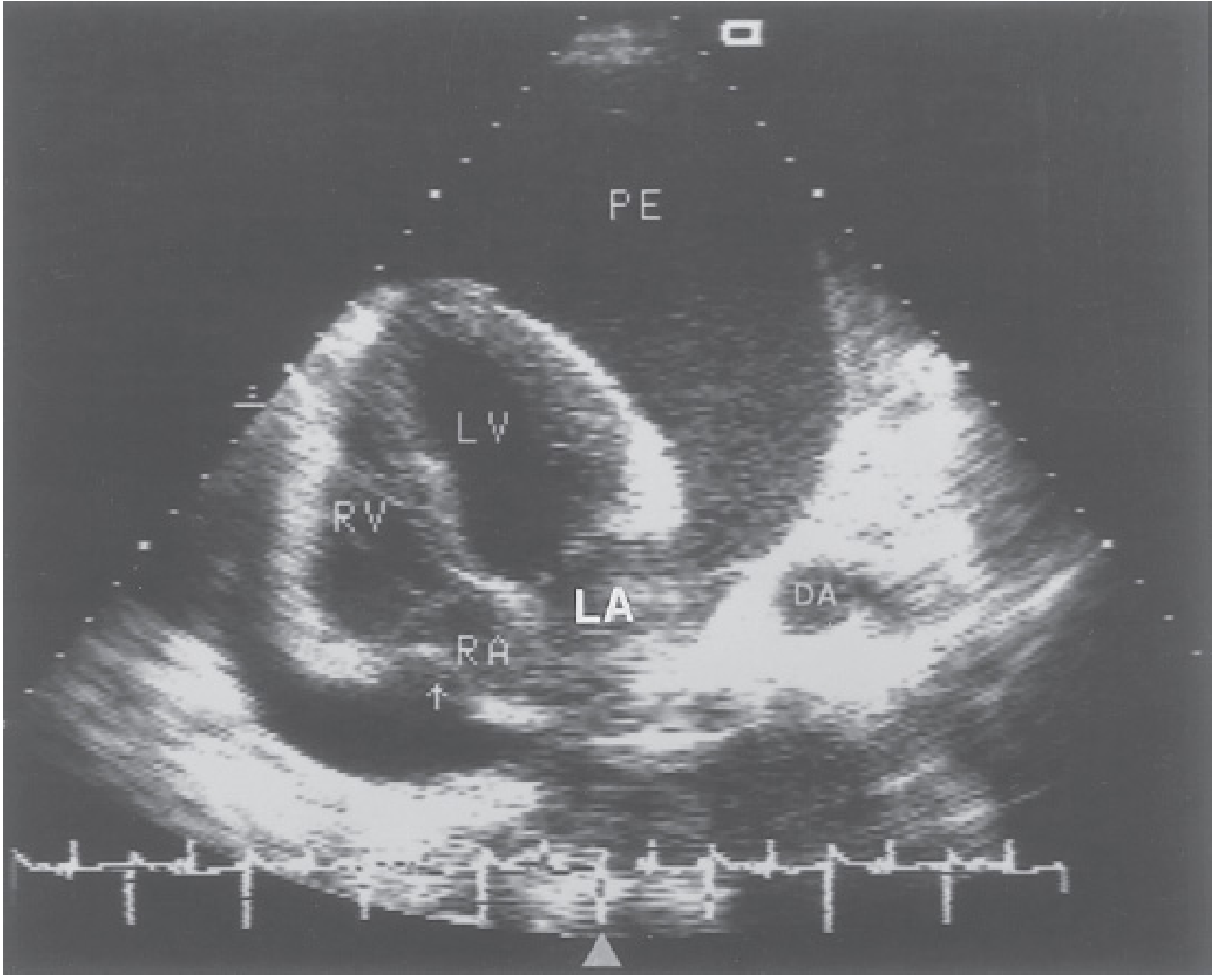
4. CT
- More accurate than echo for loculated effusions (especially anterior)
- Quantifies volume more precisely (trace the effusion on serial slices)
- Characterizes effusion: simple/transudative (near water density, -10 to +10 HU) vs complex/exudative/hemorrhagic (higher HU)
- Detects pericardial thickening, enhancement, calcification (best modality for calcification)
- Identifies associated mediastinal or pulmonary pathology
5. MRI
- Best for tissue characterization:
- Transudative/exudative (no debris): T1 hypointense, T2/FLAIR/SSFP hyperintense
- Proteinaceous or hemorrhagic: T1 hyperintense
- Fibrinous strands visible on GRE/SSFP
- Late gadolinium enhancement of pericardium = active inflammation
- Can identify associated pericardial thickening and assess for constriction
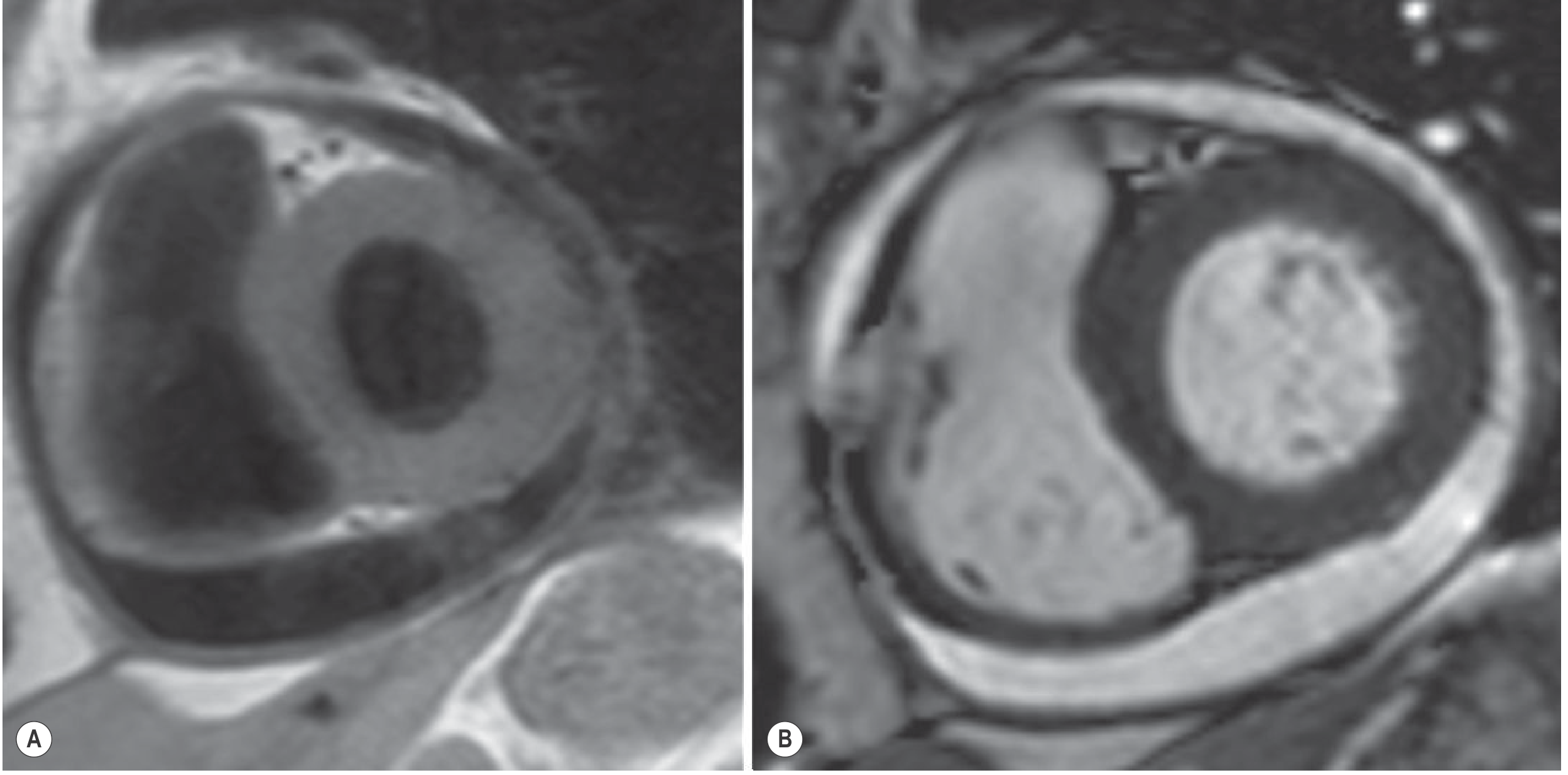
6. Pericardial Fluid Analysis
When pericardiocentesis is performed:
| Finding | Interpretation |
|---|---|
| Exudate (LDH, protein criteria) | Infection, malignancy, autoimmune |
| Transudate | CHF, cirrhosis, renal failure |
| Bloody | Trauma, aortic dissection, malignancy, post-cardiac surgery |
| Cytology positive | ~85% sensitivity for malignant effusion |
| AFB smear/culture | TB pericarditis |
| ADA elevated | TB pericarditis (>45 U/L suggestive) |
Management
Step 1: Assess hemodynamic stability
Is tamponade present?
- Hemodynamically unstable (hypotension + JVD + tachycardia + pulsus paradoxus) → emergency pericardiocentesis
- IV fluids and inotropes can temporize hypotension but must not delay drainage
Step 2: Drainage - Pericardiocentesis
Indications:
- Cardiac tamponade (emergent)
- Large symptomatic effusion (>20 mm on echo)
- Suspected purulent or tuberculous pericarditis
- Diagnostic (unexplained effusion, suspect malignancy)
- Persistent moderate-large effusion not responding to medical therapy
Technique:
- Echocardiography-guided (standard of care; reduces complications)
- Subxiphoid approach (most common) or apical
- Indwelling catheter for 2-3 days allows complete drainage and reduces recurrence
- Fluid should be sent for cytology, culture, ADA, protein, LDH, glucose
Contraindications/Caution:
- Loculated or posterior effusions (may need surgical drainage)
- Coagulopathy (correct first if possible)
- Aortic dissection (drainage will accelerate death - needs surgery)
When open drainage is preferred:
- Loculated effusion
- Blood clots (e.g., post-cardiac surgery)
- Bacterial/tuberculous pericarditis (thick exudate may block needle)
- Recurrent malignant effusion (consider pericardiectomy/pericardial window)
Step 3: Treat the underlying cause
| Cause | Specific Treatment |
|---|---|
| Idiopathic / viral | NSAIDs + colchicine (0.5 mg twice daily x 3 months) |
| Bacterial | Antibiotics + urgent drainage |
| Tuberculous | Anti-TB therapy (INH, rifampicin, PZA, ethambutol); steroids may reduce constriction |
| Malignant | Pericardiocentesis + treat primary cancer; pericardial window for recurrence; intrapericardial chemotherapy/sclerosants |
| Uremic | Intensify dialysis; NSAIDs |
| Autoimmune/inflammatory | NSAIDs + colchicine; steroids for refractory cases (caution: increase relapse) |
| Post-MI / Dressler | NSAIDs + colchicine; aspirin preferred post-MI |
| Radiation-induced | NSAIDs; if constriction develops → pericardiectomy |
| CHF/renal/hepatic | Treat underlying condition; diuretics |
Malignant Pericardial Effusion (special consideration)
-
Fluid cytology positive in ~85% of cases
-
Complete drainage with indwelling catheter 2-3 days is treatment of choice
-
Colchicine 0.6 mg twice daily after drainage reduces recurrence
-
Pericardial window (surgical or percutaneous balloon) for recurrent effusions
-
Goldman-Cecil Medicine; Mulholland & Greenfield's Surgery, pp. 4572-4575
Cardiac Tamponade - Summary
| Feature | Detail |
|---|---|
| Beck's Triad | Hypotension + JVD + muffled heart sounds |
| Most sensitive sign | Tachycardia, JVD (each ~100% sensitivity) |
| Most specific bedside sign | Pulsus paradoxus >10 mmHg |
| ECG | Electrical alternans (pathognomonic); low voltage; tachycardia |
| CXR | Water-bottle heart; rapid size increase |
| Echo | RA systolic collapse + RV diastolic collapse + IVC plethora + respiratory variation |
| Treatment | Pericardiocentesis (do not delay for IV fluids alone) |
| y-descent | Lost (tricuspid cannot open freely - blood only enters when blood leaves) |
| Pulsus paradoxus absent in | LV dysfunction, ASD, IPPV, aortic regurgitation |
Complication: Constrictive Pericarditis
Chronic pericardial inflammation may heal with fibrosis and calcification producing constrictive pericarditis - a late complication of pericardial effusion. Common causes: radiation (most common in developed world now), post-cardiac surgery, idiopathic; tuberculosis was previously dominant. Features include:
- Kussmaul's sign (paradoxical JVP rise on inspiration)
- Pericardial knock (loud S3 = abrupt cessation of diastolic filling)
- Ascites, hepatomegaly, peripheral edema (right heart failure picture)
- Treatment: pericardiectomy
Quick Reference Classification
| Size (echo) | Management |
|---|---|
| Small (<10 mm), asymptomatic | Observe; treat underlying cause |
| Moderate (10-20 mm), symptomatic | Medical ± pericardiocentesis |
| Large (>20 mm) or tamponade | Pericardiocentesis |
| Recurrent / malignant | Pericardial window / pericardiectomy |
2025 ACC Guidance (PMID 40767817): The 2025 ACC Expert Consensus Statement on Pericarditis/Pericardial Disease now emphasizes multimodality imaging (echo, CT, CMR), risk stratification for high-risk features (fever >38°C, subacute onset, large effusion, troponin rise, immunosuppression), and the role of colchicine as standard adjunctive therapy. A 2023 review on chronic pericardial effusion (PMID 36773704) highlights that idiopathic cause remains most common chronically, with colchicine and pericardiectomy as the main strategies for refractory cases.
This is a shared conversation. Sign in to Orris to start your own chat.