1 A 50 year old male patient, factory worker by profession with low socioeconomic status presented to medicine clinic with history of evening rise fever, weight loss, night sweats, fatigue and productive cough. X-ray revealed patchy and cavitary lesions in both lungs. Laboratory findings had increased ESR and sputum was showing acid fast bacilli. (A) Give your probable diagnosis (B) Describe etiopathogenesis of your probable diagnosis (C) Describe gross and microscopic morphology
(A) Probable Diagnosis
(B) Etiopathogenesis
The Organism
Transmission
Primary Infection and Immune Evasion
- Inhibiting phagolysosome fusion (via lipid-rich cell wall)
- Scavenging reactive oxygen and nitrogen intermediates
- Producing cord factor (trehalose dimycolate), which disrupts mitochondrial membranes
Cell-Mediated Immunity and Granuloma Formation (the Central Pathogenetic Event)
- Mycobacterial antigens are processed by dendritic cells and presented via MHC II to CD4+ T helper cells (TH1 subset).
- TH1 cells secrete IFN-γ, which activates macrophages, enhancing their bactericidal capacity (increased lysosomal enzymes, nitric oxide production).
- Activated macrophages transform into epithelioid cells and fuse to form Langhans giant cells.
- These cells, together with lymphocytes, fibroblasts, and a rim of fibrous tissue, form the tuberculous granuloma (tubercle).
- The center of the granuloma undergoes caseous necrosis - a type of coagulative necrosis with a cheese-like consistency, caused by delayed-type hypersensitivity (DTH) reactions mediated by TNF-α and lymphotoxin.
In approximately 95% of immunocompetent individuals, cell-mediated immunity is sufficient to contain (but not eradicate) the infection. The Ghon focus heals with fibrosis and calcification (Ranke complex). Viable bacilli remain dormant in the necrotic material for decades.
Reactivation (Secondary TB) - The Mechanism in This Patient
- Intense tissue inflammation and rapid granuloma formation
- Caseation expands and liquefies
- Liquefied caseum is expelled into airways, creating cavities - the hallmark of secondary TB
- Cavitary walls are poorly lined, making them an ideal reservoir for bacterial multiplication
- Bacilli are shed into sputum - the patient becomes infectious
(C) Gross and Microscopic Morphology
Gross Morphology
Primary TB (Ghon Complex)
- A 1.0-1.5 cm gray-white area of consolidation (Ghon focus) appears in the subpleural lower upper lobe or upper lower lobe.
- The center shows caseous necrosis - soft, cheese-like, yellow-white material.
- Regional hilar lymph nodes are enlarged with central caseation.
- Together, the parenchymal focus + caseating lymph nodes = Ghon complex (visible on chest X-ray as a Ranke complex after calcification).
Secondary TB (this patient's lesion)
- Location: Apex of one or both upper lobes (the classic location).
- Initial lesion: Small focus of consolidation, <2 cm, sharply circumscribed, firm, gray-to-yellow with central caseation and peripheral fibrosis.
- Cavitation: As disease progresses, caseous material liquefies and is expelled into the bronchus, forming a ragged, irregular cavity lined by soft caseous debris. The cavity is poorly walled off by fibrous tissue.
- Size of cavities: Can range from 2 cm to large, confluent spaces several centimeters across.
- Bilateral involvement (as in this patient): Spread via bronchi leads to bilateral patchy consolidation - the "Simon foci" at the apices with lower lobe involvement via bronchogenic spread.
- In chronic cases: Fibrous scarring, calcification, and architectural distortion of the lung.
- Miliary TB: If massive hematogenous spread occurs, 1-2 mm millet seed-like granulomas are seen throughout both lungs (and other organs).
Microscopic Morphology
| Zone | Component | Appearance on H&E |
|---|---|---|
| Center | Caseous necrosis | Amorphous, granular, acellular eosinophilic debris - "cheesy" material; no preserved cellular architecture |
| Middle | Epithelioid cells + Langhans giant cells | Epithelioid macrophages with abundant pale pink cytoplasm and elongated ("footprint-shaped") nuclei; Langhans giant cells have horseshoe/ring arrangement of nuclei at the periphery |
| Periphery | Lymphocytes + fibroblasts | Rim of CD4+ T lymphocytes and plasma cells; outer fibrous capsule in older lesions |
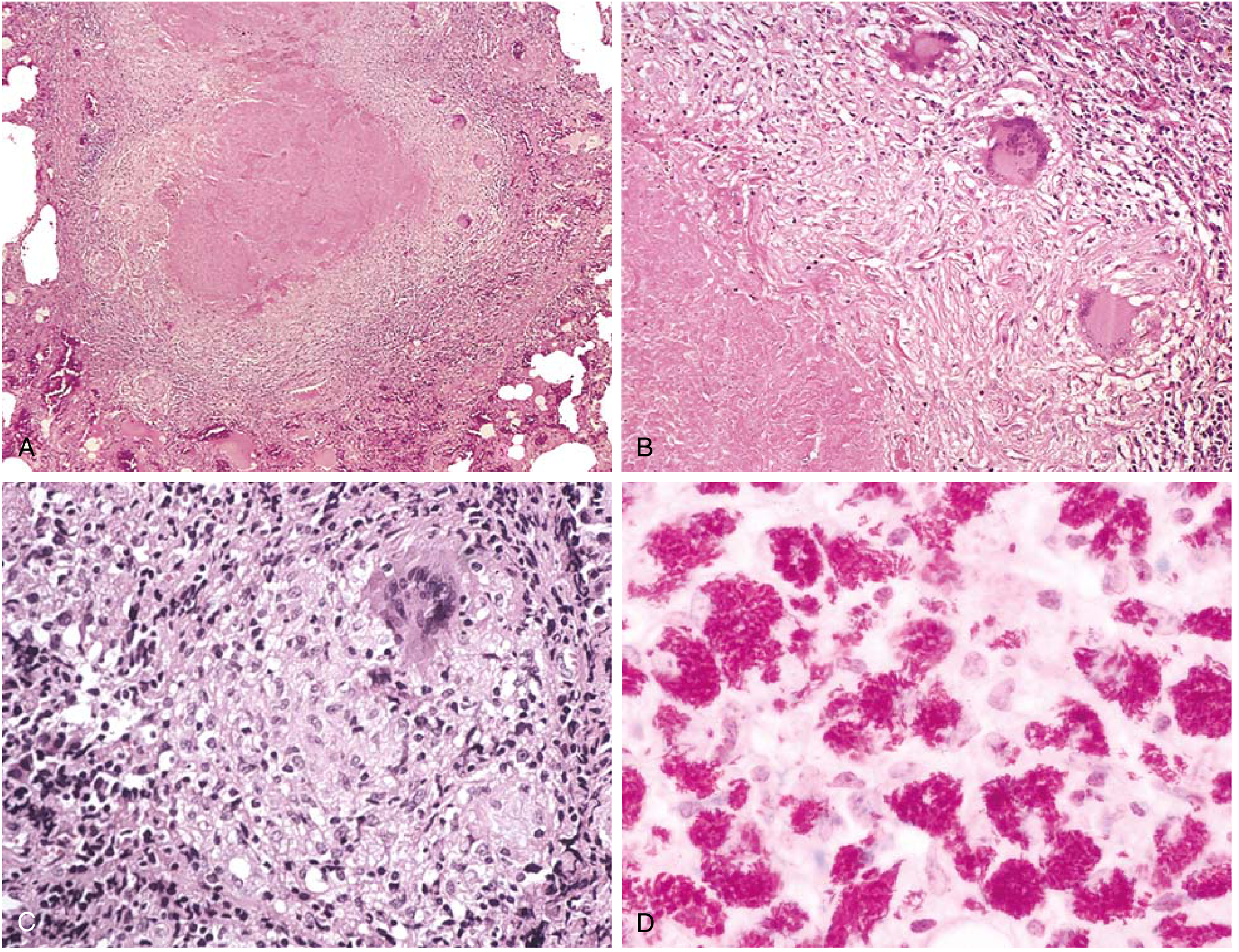
Key Microscopic Points:
- AFB staining (Ziehl-Neelsen / Auramine-rhodamine): Bacilli appear as red (ZN) or fluorescent (auramine) beaded rods on a blue-green background. They are best seen in early exudative and caseous phases; difficult to find in late fibrocalcific stages.
- Exudative phase: Early lesion - serous or fibrinous exudate with macrophages and occasional PMNs; no granuloma yet.
- Productive (granulomatous) phase: Classic caseating granuloma as described above.
- Healing phase: Progressive fibrosis, hyalinization, calcification, and sometimes ossification of the caseous material.
- In immunocompromised patients (severe HIV, etc.): Granulomas are absent or poorly formed; instead, sheets of foamy/lipid-laden macrophages packed with AFB are seen - a "non-reactive TB" pattern.
| Feature | Finding in This Case |
|---|---|
| Causative organism | Mycobacterium tuberculosis |
| Type | Secondary (reactivation) pulmonary TB |
| Key virulence factor | Cord factor (trehalose dimycolate), mycolic acid-rich cell wall |
| Immune response | TH1-mediated, IFN-γ driven, macrophage activation |
| Central pathologic lesion | Caseating granuloma with Langhans giant cells |
| Gross hallmark | Bilateral apical cavitary lesions |
| Microscopic hallmark | Caseating granuloma (epithelioid cells + Langhans giant cells + caseous center + lymphocytic rim) |
| Diagnostic stain | Ziehl-Neelsen (acid-fast bacilli appear red) |
1 A 50 year old male patient, factory worker by profession with low socioeconomic status presented to medicine clinic with history of evening rise fever, weight loss, night sweats, fatigue and productive cough. X-ray revealed patchy and cavitary lesions in both lungs. Laboratory findings had increased ESR and sputum was showing acid fast bacilli. (A) Give your probable diagnosis (B) Describe etiopathogenesis of your probable diagnosis (C) Describe gross and microscopic morphology
Secondary (Reactivation) Pulmonary Tuberculosis
(A) Probable Diagnosis
| Clue | Significance |
|---|---|
| 50-year-old male, factory worker, low SES | Classic risk profile - poverty, crowding, possible dust exposure |
| Evening rise of fever, night sweats, weight loss, fatigue | Constitutional "B-symptoms" of chronic granulomatous infection |
| Productive cough | Active bronchopulmonary disease with airway involvement |
| Bilateral patchy and cavitary lesions on X-ray | Pathognomonic of secondary/reactivation TB - cavitation from liquefied caseum expelled into airways |
| Elevated ESR | Non-specific but consistent with chronic inflammatory/infectious disease |
| Acid-fast bacilli (AFB) in sputum | Definitive microbiological confirmation of active TB |
(B) Etiopathogenesis
The Causative Organism
- Confer resistance to decolorization by acid-alcohol (basis of Ziehl-Neelsen/AFB staining)
- Enable survival inside phagolysosomes
- Drive granuloma formation and delayed-type hypersensitivity (DTH)
- Make the organism extremely slow-growing (doubling time ~15-20 hours)
Step 1: Transmission
Step 2: Initial Alveolar Deposition and Macrophage Uptake
- Inhibits phagosome-lysosome fusion - mycobacterial cell wall components (lipoarabinomannan/LAM) block the acidification of the phagosome
- Survives and replicates inside the macrophage phagosome
- Inhibits apoptosis of the host macrophage, preventing an early immune alert
- Slowly multiplies over 2-3 weeks, destroying the macrophage
Step 3: Development of Cell-Mediated Immunity (2-8 weeks)
- Mycobacterial antigens are processed and presented by dendritic cells via MHC class II to naive CD4+ T helper cells
- Under the influence of IL-12 (from activated macrophages), CD4+ T cells differentiate into TH1 cells
- TH1 cells secrete IFN-γ and TNF-α, which:
- Activate macrophages to upregulate their killing mechanisms (reactive oxygen species, nitric oxide via iNOS)
- Overcome the phagosome fusion block
- Stimulate macrophage transformation into epithelioid cells
- This is detectable at 2-8 weeks as conversion of the Mantoux/PPD test or IGRA (IFN-γ release assay)
Step 4: Granuloma Formation - The Hallmark Pathologic Response
- Macrophages transform into pale, elongated epithelioid cells (abundant cytoplasm, elongated "footprint-shaped" nuclei)
- Multiple epithelioid cells fuse to form Langhans giant cells (nuclei arranged in a horseshoe/peripheral ring pattern)
- A surrounding rim of CD4+ lymphocytes, plasma cells, and fibroblasts encircles the lesion
- The center undergoes caseous necrosis - an amorphous, cheese-like coagulative necrosis driven by DTH reactions and cytotoxic T cells; it is unique to TB (and a few other mycobacterial/fungal infections)
- The outer fibrous wall progressively calcifies in healed lesions
Step 5: Primary Infection Outcome - Latency in ~95%
- The Ghon focus (primary lung lesion) heals with fibrosis and calcification
- The Ghon complex (Ghon focus + caseating hilar lymph nodes) calcifies into the Ranke complex visible on X-ray
- Viable bacilli remain dormant within healed granulomas for decades, held in check by immune surveillance
- The host is infected but has no active disease and is non-infectious
Step 6: Reactivation (Secondary TB) - The Mechanism in This Patient
- Malnutrition (low SES)
- Chronic fatigue/debility
- Possible silica dust exposure (silicosis is a major risk factor for TB)
- Aging (declining immune surveillance)
- Other: diabetes, alcohol use
- Dormant bacilli in apical foci resume replication
- Rapid granuloma formation due to pre-existing sensitization
- Central caseation enlarges and liquefies (due to enzymatic degradation by macrophage proteases and DTH-mediated necrosis)
- The liquefied caseum erodes into an airway/bronchus and is expelled - creating the classic CAVITY
- The cavity wall is irregular, poorly lined by fibrous tissue, providing an ideal warm, oxygenated, nutrient-rich environment for massive bacillary multiplication
- Bacilli shed into sputum make the patient infectious
- Erosion of blood vessels causes hemoptysis
- Bronchogenic spread to lower lobes and the opposite lung creates bilateral patchy consolidation
Key distinction: Unlike primary TB, regional lymphadenopathy is less prominent in secondary TB because the rapid hypersensitivity response walls off the focus before lymphatic spread occurs significantly.
(C) Gross and Microscopic Morphology
Gross Morphology
Primary TB (Ghon Complex)
- Ghon focus: A 1.0-1.5 cm, gray-white, subpleural area of consolidation in the lower part of the upper lobe or upper part of the lower lobe; center shows soft, yellow-white caseous necrosis
- Hilar lymph nodes: Enlarged, with central caseous necrosis
- Ghon complex = Ghon focus + caseating hilar nodes (together); undergoes progressive fibrosis and calcification to form the Ranke complex
- In 95% of cases, this is the endpoint - disease is controlled
Secondary TB (This Patient's Disease)
- Location: Apex of one or both upper lobes (classic apical localization)
- Initial apical lesion: Small (<2 cm), sharply circumscribed, firm, gray-to-yellow area with central caseation and peripheral fibrosis; can be felt as a firm nodule on sectioning
- Progressive disease - Cavitation:
- As caseation expands and liquefies, it erodes into a bronchus
- A ragged, irregular cavity forms, lined by soft caseous material, poorly walled off by fibrous tissue
- Cavity size ranges from 2 cm to large confluent spaces several centimeters across
- Cavity walls are thick initially; with treatment, they can collapse and fibrose
- Bilateral patchy lesions (as in this patient): Bronchogenic spread deposits infected caseous material in lower lobes and the contralateral lung, producing patchy areas of consolidation, new caseating foci, and smaller satellite cavities
- Spread patterns in progressive disease:
- Bronchogenic spread - most common; causes bilateral lower lobe involvement
- Lymphatic spread - hilar lymphadenopathy (less prominent than primary)
- Hematogenous spread - miliary TB: 1-2 mm millet seed-like granulomas disseminated throughout both lungs and other organs
- Direct extension - pleuritis, pericarditis, empyema
- Healed lesions: Progressive fibrous encapsulation → fibrocalcific scars → architectural distortion of pulmonary parenchyma
Microscopic Morphology
- Amorphous, granular, acellular eosinophilic debris on H&E
- Complete loss of normal tissue architecture (unlike coagulative necrosis where cell outlines are preserved)
- Cheese-like consistency grossly ("caseous" = cheese-like in Latin)
- AFB may be demonstrable here in early/active lesions by Ziehl-Neelsen stain
- Epithelioid cells: Transformed, activated macrophages with abundant pale-pink cytoplasm and elongated, "footprint-shaped" or "shoe sole-shaped" nuclei - the most characteristic cell of TB
- Langhans giant cells: Multinucleated giant cells formed by fusion of epithelioid macrophages; nuclei arranged in a horseshoe or peripheral ring at the cell margin (distinguishes Langhans from foreign-body giant cells which have randomly scattered nuclei)
- Foamy macrophages: Lipid-laden macrophages in the interlayers of the granuloma, providing nutrition to inflammatory cells
- Dense rim of CD4+ T lymphocytes and plasma cells
- Fibroblasts and collagen fibers forming the outer capsule
- Thickens with healing to form complete fibrous encapsulation
- Exudative phase (earliest): Serous or fibrinous exudate + macrophages ± PMNs; no granuloma yet; bacilli plentiful
- Productive/granulomatous phase: Classic caseating granuloma as above
- Healing/fibrocalcific phase: Fibrosis, hyalinization, calcification, and sometimes ossification; bacilli rarely demonstrable at this stage
- Immunocompromised patients (non-reactive TB): Granulomas are absent or poorly formed; instead, sheets of foamy/lipid-laden macrophages packed with AFB are seen - a "non-reactive" or "anergic" pattern
- Ziehl-Neelsen (ZN) stain / Kinyoun stain: AFB appear as bright red, beaded rods against a blue counterstain background
- Auramine-rhodamine fluorescence stain: AFB fluoresce bright yellow-orange; more sensitive than ZN
- PAS stain: Highlights fungal organisms to exclude fungi in differential diagnosis of granulomas
Summary Overview
| Feature | Detail |
|---|---|
| Diagnosis | Secondary (reactivation) pulmonary TB |
| Causative organism | Mycobacterium tuberculosis - acid-fast bacillus |
| Transmission | Airborne droplet nuclei (person-to-person) |
| Key immune response | TH1-mediated, IFN-γ driven macrophage activation |
| Central pathologic reaction | Caseating granuloma (delayed-type hypersensitivity + cell-mediated immunity) |
| Gross hallmark | Bilateral apical cavitary lesions (ragged, caseous-lined cavities) |
| Microscopic hallmark | Caseating granuloma: caseous center + epithelioid cells + Langhans giant cells + lymphocytic rim |
| Key cell | Epithelioid cell (activated macrophage) + Langhans giant cell |
| AFB stain appearance | Bright red beaded rods (ZN stain) |
| Latency mechanism | Viable dormant bacilli in healed granulomas; hypoxia triggers latency regulators |
| Reactivation trigger | Immune suppression (malnutrition, silicosis, HIV, aging, diabetes, alcohol) |
Define regeneration and repair
Regeneration and Repair
Definitions
Repair
"Repair of damaged tissues occurs by two processes: regeneration, which restores normal cells, and scarring, the deposition of connective tissue."
- Robbins, Cotran & Kumar Pathologic Basis of Disease
Regeneration
- Proliferation of differentiated cells that survive the injury and retain the capacity to divide (e.g., hepatocytes regenerating after partial hepatectomy or liver injury)
- Differentiation and maturation of tissue stem cells (e.g., intestinal stem cells at the base of crypts continuously regenerating the gut epithelium; hematopoietic stem cells replenishing blood cells)
- The injured tissue must have proliferative capacity, AND
- The structural scaffolding (extracellular matrix/basement membrane) must remain intact to guide the regenerating cells back into their correct architecture
Repair by Scar Formation
"If the injured tissue is not capable of proliferation, or if the structural framework is damaged and cannot support regeneration, repair occurs by replacement with connective tissue and scar formation."
- Robbins & Kumar Basic Pathology
Classification of Tissues by Regenerative Capacity
| Type | Characteristics | Examples | Regenerative Capacity |
|---|---|---|---|
| Labile (Continuously Dividing) | Continuously lost and replaced from stem cell pools; cells always in cycle | Skin epidermis, oral/GI/respiratory/urogenital epithelium, hematopoietic cells | High - readily regenerate after injury as long as stem cells are preserved |
| Stable (Quiescent) | Normally quiescent (G0 phase); can re-enter cell cycle in response to injury | Liver, kidney, pancreas, endothelial cells, fibroblasts, smooth muscle cells | Moderate - can divide when stimulated; liver has exceptional regenerative capacity; most other stable organs have limited regeneration |
| Permanent (Non-dividing) | Terminally differentiated; cannot re-enter cell cycle after injury | Neurons, cardiac muscle cells, skeletal muscle | None (or negligible) - injury leads to irreversible loss and scar formation |
Key Differences: Regeneration vs. Repair by Scarring
| Feature | Regeneration | Repair (Scar Formation) |
|---|---|---|
| End result | Normal tissue restored | Fibrous scar - structurally stable but functionally inferior |
| Cell type replacing lost tissue | Same parenchymal cell type | Fibroblasts/connective tissue |
| ECM scaffold | Must be intact | Framework may be destroyed |
| Tissue type | Labile and some stable tissues | Permanent tissues; also when scaffold is gone |
| Function | Fully restored | Partially/not restored |
| Example | Liver regeneration after hepatectomy; skin abrasion healing | Myocardial infarction healing; CNS injury |
Classic Example: Liver Regeneration
- Surviving hepatocytes are stimulated by cytokines (TNF-α, IL-6) and growth factors (HGF - hepatocyte growth factor; EGF)
- Quiescent hepatocytes re-enter the cell cycle and proliferate
- If the injury is severe enough to exhaust the hepatocyte proliferative pool, liver stem cells (oval cells/ductal progenitors) are activated
- Original liver architecture is fully restored if the ECM scaffold remains intact
Practical Significance
- Skin wounds heal by regeneration of the epithelium over a connective tissue base (scar in dermis)
- Bone fractures heal by regeneration if properly aligned; misaligned fractures heal with fibrous/cartilaginous callus
- Myocardial infarction heals only by scarring - permanent loss of contractile tissue
- Peripheral nerves can regenerate (Schwann cells and intact endoneurial tubes guide axonal regrowth); CNS neurons cannot
Demonstration of amyloid as 2 mark answer
Demonstration of Amyloid
- Under ordinary (bright-field) light: amyloid deposits appear pink or red
- Under polarized light: amyloid shows pathognomonic apple-green (yellow-green) birefringence - this is the single most specific diagnostic feature
- This reaction is shared by all chemical forms of amyloid and is caused by the cross-β-pleated sheet configuration of amyloid fibrils, which bind Congo red dye in a regular, repeating pattern
- Electron microscopy: Reveals characteristic non-branching, randomly oriented fibrils (7.5-10 nm diameter)
- Immunohistochemistry: Identifies specific amyloid types (AA, TTR)
- Mass spectroscopy / protein sequencing: Definitive subtyping, especially for AL amyloid
Key exam point: Congo red stain + apple-green birefringence under polarized light = definitive demonstration of amyloid.
Pediatric tumors example
Pediatric Tumors - Examples
Special Tumorlike Lesions (Unique to Pediatrics)
- Heterotopia (Choristoma): Microscopically normal cells/tissues present in an abnormal location (e.g., ectopic pancreatic tissue in the stomach wall; adrenal cells found in the kidney)
- Hamartoma: Excessive but focal overgrowth of cells native to the organ, but with disordered architecture (e.g., hemangiomas, rhabdomyomas of the heart) - the line between hamartoma and a benign neoplasm is often blurred
A. Benign Tumors of Childhood
1. Hemangioma
- Most common neoplasm of infancy
- Located on skin (face, scalp) - irregular, erythematous to violaceous, flat to nodular lesions
- Both cavernous and capillary types; appear more cellular than adult hemangiomas
- Many spontaneously regress after birth
2. Lymphangioma
- Benign tumor of lymphatic channels
- Can occur in skin, deep soft tissues, and viscera
3. Teratoma
- Arise from pluripotent germ cells containing tissues from all three germ layers (ectoderm, mesoderm, endoderm)
- Most common site: sacrococcygeal region (Fig. 4.37 below - note the massive size of the lesion relative to the infant)
- The majority (>90%) in infants are benign (mature teratoma); malignant potential increases with age
- Also occur in gonads, mediastinum, and retroperitoneum
B. Malignant Tumors of Childhood
Common Malignant Neoplasms by Age Group
| 0-4 Years | 5-9 Years | 10-14 Years |
|---|---|---|
| Leukemia | Leukemia | Leukemia |
| Retinoblastoma | Retinoblastoma | Hepatocellular carcinoma |
| Neuroblastoma | Neuroblastoma | Soft tissue sarcoma |
| Wilms tumor | Hepatocellular carcinoma | Osteosarcoma |
| Hepatoblastoma | Soft tissue sarcoma | Thyroid carcinoma |
| Rhabdomyosarcoma | Ewing sarcoma | Hodgkin lymphoma |
| Teratomas | CNS tumors | |
| CNS tumors | Lymphoma |
- Neuroblastoma
- Lymphoma
- Rhabdomyosarcoma (most common soft tissue sarcoma of childhood)
- Ewing sarcoma
- Medulloblastoma
- Retinoblastoma
- Wilms tumor (some cases)
1. Neuroblastoma
- Arises from primitive neural crest cells of sympathetic ganglia and adrenal medulla
- Second most common solid malignancy of childhood after brain tumors (7-10% of all pediatric neoplasms; up to 50% of malignancies in infancy)
- Most arise in the adrenal medulla or paraspinal sympathetic ganglia
- Unique features:
- Spontaneous regression (especially in infants <1 year)
- Spontaneous or therapy-induced maturation into ganglioneuroma (benign)
- 1-2% are familial (autosomal dominant; ALK gene mutations)
- Histology: Small round blue cells with Homer-Wright pseudorosettes (tumor cells arranged around a central neuropil core)
- Markers: Urinary catecholamines (VMA, HVA) elevated; N-MYC amplification = poor prognosis
2. Wilms Tumor (Nephroblastoma)
- Most common primary renal tumor of childhood; peak age 3-4 years
- Associated with WAGR syndrome (Wilms tumor, Aniridia, Genitourinary anomalies, mental Retardation) and WT1 gene mutations on chromosome 11p13
- Histology: Classic triphasic pattern - blastemal cells + stromal cells + epithelial (tubular) cells replicating embryonic kidney development
- Generally good prognosis with multimodal therapy (surgery + chemotherapy ± radiation)
3. Retinoblastoma
- Most common intraocular malignancy of childhood
- Due to RB1 tumor suppressor gene mutation (chromosome 13q14) - the prototype of the "two-hit hypothesis" (Knudson)
- Can be hereditary (bilateral, autosomal dominant) or sporadic (unilateral)
- Histology: Flexner-Wintersteiner rosettes (tumor cells around a central lumen)
4. Rhabdomyosarcoma
- Most common soft tissue sarcoma of childhood
- Arises from primitive mesenchymal cells committed to skeletal muscle differentiation
- Common sites: head/neck, genitourinary tract, extremities
- Subtypes: embryonal (most common, better prognosis) and alveolar (worse prognosis, PAX-FOXO1 fusion)
5. Ewing Sarcoma
- Malignant bone tumor of childhood/adolescence; affects diaphysis of long bones
- Characterized by t(11;22) translocation - EWS-FLI1 fusion gene
- "Onion-skin" periosteal reaction on X-ray
- Histology: uniform small round blue cells
6. Hepatoblastoma
- Most common primary liver tumor in children <5 years
- Associated with familial adenomatous polyposis (APC mutations); elevated AFP
- Histology: embryonal and fetal hepatocytic patterns
7. Medulloblastoma
- Most common malignant CNS tumor of childhood
- Located in cerebellum; small round blue cells with Homer-Wright rosettes
- Frequently metastasizes via CSF pathways
Key Distinguishing Features of Pediatric vs. Adult Tumors
| Feature | Pediatric Tumors | Adult Tumors |
|---|---|---|
| Origin | Often from embryonal/primitive cells | Usually from differentiated epithelium |
| Histology | Small round blue cells; immature elements | Carcinomas predominate |
| Prognosis | Often better - can mature or spontaneously regress | Generally worse |
| Genetics | Tumor suppressor mutations (WT1, RB1); translocations | Accumulated somatic mutations |
| Response to chemotherapy | Generally more sensitive | Variable |
Pediatric tumors 2 examples
Pediatric Tumors - Two Classic Examples
1. Neuroblastoma
- Second most common solid malignancy of childhood after brain tumors
- Accounts for 7-10% of all pediatric neoplasms
- Up to 50% of all malignancies diagnosed in infancy
- Most common in children under 2 years
- Most are sporadic; 1-2% are familial (autosomal dominant; ALK gene mutations)
- N-MYC amplification = marker of poor prognosis
- Somatic gain-of-function ALK mutations in 8-10% of sporadic cases
- Spontaneous regression - particularly in infants <1 year, even with metastatic disease
- Maturation - can differentiate into benign ganglioneuroma (spontaneously or with therapy)
- Small, round, blue cells - the classic "small round blue cell tumor"
- Homer-Wright pseudorosettes - tumor cells arranged in a circle around a central tangle of neuropil fibrils (no lumen) - pathognomonic
- Stroma may show Schwannian differentiation in maturing tumors
- Abdominal mass (most common presentation)
- Elevated urinary catecholamines: VMA (vanillylmandelic acid) and HVA (homovanillic acid)
- "Raccoon eyes" (periorbital ecchymosis) from orbital metastasis
- Opsoclonus-myoclonus syndrome (paraneoplastic)
- Infants <1 year: excellent (even with metastases - spontaneous regression common)
- Older children with N-MYC amplification: poor prognosis
2. Wilms Tumor (Nephroblastoma)
- Most common primary renal tumor of childhood
- Peak incidence: 3-4 years of age
- Accounts for ~6% of all pediatric cancers
- WT1 gene (Wilms Tumor 1) - tumor suppressor on chromosome 11p13; loss of function leads to tumor
- Associated with:
- WAGR syndrome - Wilms tumor, Aniridia, Genitourinary anomalies, mental Retardation (WT1 deletion)
- Beckwith-Wiedemann syndrome - overgrowth syndrome; WT2 locus on 11p15
- Denys-Drash syndrome - gonadal dysgenesis + nephropathy + Wilms tumor
| Component | Appearance |
|---|---|
| Blastemal cells | Sheets of small, round, primitive blue cells with scant cytoplasm |
| Stromal cells | Spindle-shaped cells; may show heterologous differentiation (cartilage, bone, fat) |
| Epithelial cells | Abortive tubules and glomeruloid structures mimicking developing kidney |
- Favorable histology (majority): Classic triphasic pattern - good prognosis
- Unfavorable/Anaplastic histology (~5%): Nuclear anaplasia present - poor prognosis; associated with TP53 mutations
- Large, solitary, well-circumscribed mass; pseudocapsule separating it from residual kidney
- Cut surface: soft, homogeneous, gray-white with areas of hemorrhage and necrosis
- Large abdominal mass (often discovered incidentally by parents/bathing)
- Abdominal pain, hematuria, hypertension
- Rarely ruptures spontaneously (but manipulation during examination can cause rupture → peritoneal seeding)
Quick Comparison
| Feature | Neuroblastoma | Wilms Tumor |
|---|---|---|
| Origin | Neural crest / adrenal medulla | Metanephric blastema / kidney |
| Peak age | <2 years | 3-4 years |
| Key gene | ALK, N-MYC | WT1 (chr 11p13) |
| Histology | Small round blue cells + Homer-Wright rosettes | Triphasic: blastemal + stromal + epithelial |
| Marker | Urinary VMA, HVA | - |
| Unique feature | Spontaneous regression / maturation | Triphasic pattern mimicking kidney development |
| Prognosis | Variable (age/N-MYC dependent) | Excellent (>90% cure rate) |
19 year old healthy young male came to blood bank on his birthday for voluntary blood donation (A) What are the criteria for a blood donor (B) Which are the mandatory pre transfusion tests (C) What are the causes of blood donor deferral (D) What is autologous blood transfusions)
Blood Donation - Complete Answer
(A) Criteria for a Blood Donor
General Eligibility Criteria
| Parameter | Requirement |
|---|---|
| Age | 18-65 years (some countries 17-65 with parental consent; upper limit may extend to 70 for repeat donors) |
| Weight | Minimum 50 kg (110 lbs) - ensures safe removal of 450 mL (one unit) without hemodynamic compromise |
| Hemoglobin | Males: ≥13.0 g/dL; Females: ≥12.5 g/dL (some standards: ≥12.0 g/dL) |
| Pulse | 60-100 beats/min; regular rhythm |
| Blood pressure | Systolic 100-180 mmHg; Diastolic 60-100 mmHg |
| Temperature | ≤37.5°C (afebrile) |
| General health | Apparently healthy with no acute illness on day of donation |
| Frequency of donation | Whole blood: maximum 4 times/year (every 3 months); minimum 12 weeks between donations |
This Patient (19-year-old healthy male)
- Age: 19 years ✓ (within range)
- Weight: presumably ≥50 kg ✓
- Hemoglobin: if ≥13.0 g/dL ✓
- Must pass medical history questionnaire and physical examination ✓
(B) Mandatory Pre-Transfusion Tests
1. Blood Grouping
- ABO typing (forward and reverse/back typing)
- Rh (D) typing - Rh positive or Rh negative
2. Infectious Disease Screening (Transfusion-Transmissible Infections - TTIs)
| Infection | Test Used | Residual Risk (per transfusion) |
|---|---|---|
| HIV-1/HIV-2 | Anti-HIV-1/HIV-2 ELISA + HIV NAT (nucleic acid testing) | ~1 : 1,800,000 |
| Hepatitis C (HCV) | Anti-HCV ELISA + HCV NAT | ~1 : 1,600,000 |
| Hepatitis B (HBV) | HBsAg + HBV NAT + Anti-HBc | ~1 : 300,000 to 1,500,000 |
| Syphilis | VDRL or RPR (nontreponemal) or treponemal test | Rarely transmitted (spirochete fragile in storage) |
| HTLV-I/II | Anti-HTLV-I/II antibody | ~1 : 3,300,000 |
| Malaria | Donor history questionnaire (endemic areas: antigen/antibody test) | Questionnaire-based |
| Chagas disease | Anti-T. cruzi antibody (tested at first donation) | Rare |
NAT (Nucleic Acid Testing) dramatically shortens the window period (the gap between infection and detectable antibody):
- HIV: window period with NAT = 9 days vs. 21 days with ELISA alone
- HCV: window period with NAT = 7 days vs. 51-58 days with ELISA
3. Pre-Transfusion Compatibility Testing (Recipient Side)
- Type and Screen - recipient ABO/Rh typing + antibody screen
- Crossmatch - major crossmatch (recipient serum + donor RBCs) to detect incompatibility
- Direct Antiglobulin Test (DAT) if clinically indicated
(C) Causes of Blood Donor Deferral
Permanent Deferral
| Condition | Reason |
|---|---|
| HIV positive / AIDS | Transfusion-transmitted infection |
| Hepatitis B or C (active or chronic) | Transfusion-transmitted infection |
| HTLV-I/II infection | Transfusion-transmitted infection |
| IV drug use (ever) | High risk for bloodborne infections |
| Men who have sex with men (MSM) - in some countries | Risk of HIV/HTLV |
| History of Chagas disease | Transfusion-transmitted infection |
| Malignancy (most cancers) | Risk of transmitting malignant cells |
| Variant Creutzfeldt-Jakob disease (vCJD) | Prion disease; no test available |
| Multiple sclerosis, epilepsy on medication | Neurological instability |
| Unexplained weight loss >3 kg in 3 months | Possible occult disease |
Temporary Deferral
| Condition | Deferral Period |
|---|---|
| Fever / acute infection | Until fully recovered (minimum 2 weeks after symptoms resolve) |
| Recent surgery (major) | 6-12 months |
| Tooth extraction / dental surgery | 24-72 hours |
| Vaccination - live attenuated (MMR, varicella, BCG) | 4 weeks after vaccination |
| Vaccination - inactivated (influenza, hepatitis B, tetanus) | 24-48 hours |
| Pregnancy | 6 months after delivery/termination |
| Breastfeeding | Until 3 months after cessation |
| Malaria risk - travel to endemic area | 3-12 months (depending on country) |
| Recent tattoo or piercing | 6-12 months (due to hepatitis B risk) |
| Blood transfusion received | 6-12 months |
| Recent sexual contact with high-risk individual | 6-12 months |
| Hemoglobin below threshold | Until Hb corrected |
| Use of certain medications (aspirin, antibiotics) | 3-7 days |
| Alcohol consumption (on day of donation) | 24-48 hours |
| Previous donation | Minimum 12 weeks (3 months) between whole blood donations |
(D) Autologous Blood Transfusion
"Autologous blood transfusion constitutes three distinct procedures: (1) preoperative autologous donation (PAD), (2) acute normovolemic hemodilution (ANH), and (3) intraoperative and postoperative blood salvage. Autologous transfusion aims to decrease the incidence and severity of complications associated with allogeneic transfusions and conserve the supply of banked blood."
- Miller's Anesthesia, 10th Edition
Three Types of Autologous Transfusion
- Patient donates their own blood 1-5 weeks before elective surgery
- Blood is stored and re-infused intraoperatively or postoperatively if needed
- AABB requirement: Hb must be ≥11 g/dL before each donation
- Repeated donations separated by at least 1 week; last donation ≥72 hours before surgery
- Risk: can cause preoperative anemia (average Hb is 1.1 g/dL lower in PAD patients)
- Use has declined significantly since the mid-1990s due to improved blood safety
- Blood withdrawn immediately before surgery in the operating room
- Circulating volume maintained with crystalloid/colloid replacement
- Stored blood (fresh, with all clotting factors intact) is re-infused at end of surgery when surgical blood loss has occurred
- Advantage: re-infused blood is fresh (superior to stored banked blood)
- Blood lost during surgery is collected from the operative field using a specialized device ("cell saver")
- Washed, filtered, and re-infused to the patient
- Widely used in cardiac surgery, orthopedic surgery, liver transplantation, vascular surgery
- Contraindicated when surgical field is contaminated (infection, malignancy)
Advantages of Autologous Transfusion
- Eliminates risk of transfusion-transmissible infections (HIV, HCV, HBV)
- Eliminates alloimmunization (antibodies against donor antigens)
- Avoids transfusion reactions (ABO/Rh incompatibility, febrile/allergic reactions)
- Ideal for patients with rare blood groups or multiple alloantibodies
- Avoids transfusion-associated GVHD
- Psychologically acceptable to patients (including some religious groups)
- Conserves allogeneic blood supply
Disadvantages / Contraindications
- Does not eliminate risk of clerical errors (wrong blood transfused)
- PAD can cause preoperative anemia - actually increases overall transfusion rate by 24%
- Autologous blood can still be contaminated during collection/storage
- Contraindications to autologous donation: active infection/bacteremia, severe aortic stenosis, unstable angina, MI or stroke within 6 months, active seizure disorder, severe cardiopulmonary disease
Summary
| Aspect | Key Point |
|---|---|
| Donor criteria | Age 18-65, weight ≥50 kg, Hb ≥13 g/dL (male), healthy, no deferral criteria |
| Mandatory tests | ABO/Rh typing + HIV, HCV, HBV, syphilis, HTLV, Chagas (NAT + serology) |
| Deferral | Permanent (HIV, hepatitis, malignancy) or temporary (infections, travel, vaccinations, pregnancy) |
| Autologous transfusion | Patient's own blood reused - three types: PAD, ANH, cell salvage; eliminates infectious and immunological risks of allogeneic transfusion |
Causes of eosinophilia
Causes of Eosinophilia
- Mild: 500-1500/μL
- Moderate: 1500-5000/μL
- Severe/Hypereosinophilia: >5000/μL (>1.5 × 10⁹/L)
1. Allergic / Atopic Disorders
- Bronchial asthma
- Allergic rhinitis (hay fever)
- Atopic dermatitis (eczema)
- Urticaria and angioedema
- Food allergies
2. Parasitic Infections
- Ascaris lumbricoides (roundworm) - especially during larval migration (Löffler syndrome)
- Strongyloides stercoralis - important: can cause fatal hyperinfection with steroids
- Hookworm (Ancylostoma, Necator)
- Toxocara canis / cati - visceral larva migrans
- Trichinella spiralis - trichinosis
- Wuchereria bancrofti - lymphatic filariasis
- Brugia malayi - tropical pulmonary eosinophilia
- Echinococcus - hydatid disease
- Schistosoma species
3. Drug Reactions
- Antibiotics: sulfonamides, ampicillin, cephalosporins, nitrofurantoin
- NSAIDs, aspirin
- Allopurinol
- Carbamazepine, phenytoin
- Gold salts
- Ranitidine, omeprazole
- Many others
4. Pulmonary Disorders with Eosinophilia
- Löffler syndrome - transient eosinophilic pulmonary infiltrates during larval migration through lungs (Ascaris)
- Tropical pulmonary eosinophilia - filarial infection causing bilateral pulmonary infiltrates + marked eosinophilia
- Chronic eosinophilic pneumonia
- Acute eosinophilic pneumonia
- Allergic bronchopulmonary aspergillosis (ABPA)
- Coccidioidomycosis
- Eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) - asthma + eosinophilia + vasculitis
5. Skin Diseases
- Atopic dermatitis
- Pemphigus / Bullous pemphigoid
- Eosinophilic folliculitis
- Eosinophilic fasciitis (Shulman syndrome)
- Eosinophilic cellulitis (Wells syndrome)
- Episodic angioedema with eosinophilia
6. Neoplasms / Malignancies
- Hodgkin lymphoma - classic association; eosinophilia is a poor prognostic sign
- Non-Hodgkin lymphoma
- Eosinophilic leukemia (primary clonal eosinophilia)
- Hypereosinophilic syndrome (HES) - idiopathic or myeloproliferative
- Lung, cervix, stomach, colon carcinomas (paraneoplastic)
- Systemic mastocytosis
7. Collagen Vascular / Autoimmune Diseases
- Eosinophilic granulomatosis with polyangiitis (Churg-Strauss)
- Polyarteritis nodosa
- Rheumatoid arthritis
- Systemic lupus erythematosus (SLE)
- Inflammatory bowel disease (Crohn's disease, ulcerative colitis)
- Sarcoidosis
8. Endocrine / Metabolic
- Addison's disease (adrenocortical insufficiency) - cortisol normally suppresses eosinophils; its absence causes eosinophilia
- Hypopituitarism
9. Gastrointestinal Disorders
- Eosinophilic esophagitis (EoE)
- Eosinophilic gastroenteritis
- Inflammatory bowel disease
10. Miscellaneous
- Hypereosinophilic syndrome (HES) - persistent eosinophilia >1500/μL for >6 months with end-organ damage and no identifiable secondary cause
- Interstitial nephritis (drug-induced)
- Transplant rejection
- Atherosclerotic embolic disease
- Eosinophilia-myalgia syndrome (L-tryptophan contamination)
- HIV / HTLV-1 infection
- Convalescent phase of acute bacterial/viral infections
- Premature birth (~75% of low-birth-weight infants show transient eosinophilia)
Summary Table
| Category | Key Examples |
|---|---|
| Allergic | Asthma, allergic rhinitis, atopic dermatitis |
| Parasitic | Strongyloides, Ascaris, hookworm, Toxocara, filariasis |
| Drugs | Sulfonamides, NSAIDs, allopurinol, anticonvulsants |
| Pulmonary | Löffler syndrome, tropical pulmonary eosinophilia, ABPA, Churg-Strauss |
| Skin | Bullous pemphigoid, eosinophilic fasciitis |
| Neoplasms | Hodgkin lymphoma, eosinophilic leukemia, HES |
| Autoimmune | Churg-Strauss, polyarteritis nodosa, IBD |
| Endocrine | Addison's disease, hypopituitarism |
| GI | Eosinophilic esophagitis, eosinophilic gastroenteritis |
| Miscellaneous | HES, transplant rejection, HIV, interstitial nephritis |
Key exam point: In the developing world, parasitic infections (especially helminths) are the leading cause. In the developed world, atopic/allergic diseases predominate. Always rule out Strongyloides before starting steroids - can cause fatal hyperinfection syndrome.
Pathogenesis of amyloidosis as 3mark
Pathogenesis of Amyloidosis
Core Mechanism - Protein Misfolding
"Amyloidosis results from abnormal folding of proteins, which assume a β-pleated sheet conformation, aggregate, and deposit as fibrils in extracellular tissues."
- Robbins & Kumar Basic Pathology
- Normally, misfolded intracellular proteins are degraded in proteasomes, and extracellular aggregates are cleared by macrophages
- In amyloidosis, these mechanisms fail - fibrillar proteins accumulate extracellularly, compressing and destroying adjacent cells
Step-by-Step Pathogenesis
Step 1: Source of Amyloidogenic Protein
- Normal proteins produced in abnormally large amounts (e.g., SAA in chronic inflammation; β2-microglobulin in dialysis patients)
- Mutant proteins that are structurally unstable and prone to misfolding (e.g., mutant transthyretin/TTR in familial amyloidosis)
Step 2: Protein Misfolding → β-Pleated Sheet Formation
- The protein loses its normal tertiary structure
- Partial unfolding exposes hydrophobic regions that self-associate
- Proteins refold into antiparallel β-pleated sheet configuration - the universal structural hallmark of all amyloid, regardless of the precursor protein
- This conformation is highly stable, resists proteolytic degradation, and is responsible for Congo red staining and apple-green birefringence
Step 3: Fibril Assembly and Extracellular Deposition
- Misfolded proteins aggregate into protofilaments → which wind together to form amyloid fibrils (4-6 protofilaments per fiber, 7.5-10 nm diameter)
- Fibrils are deposited extracellularly, initially along basement membranes
- As deposits grow, they encroach on, compress, and ultimately destroy adjacent parenchymal cells
Classification by Amyloid Precursor Protein
| Type | Precursor Protein | Clinical Setting | Trigger |
|---|---|---|---|
| AL (Amyloid Light chain) | Immunoglobulin light chains (λ > κ) | Primary amyloidosis, Multiple myeloma | Clonal plasma cell proliferation; λ chains 6× more amyloidogenic than κ |
| AA (Amyloid-Associated) | Serum Amyloid A (SAA) - an acute-phase protein | Secondary/Reactive amyloidosis (rheumatoid arthritis, TB, Crohn's, chronic infections) | Chronic inflammation → IL-6 and IL-1 stimulate liver to produce SAA → sustained elevation → fibril formation |
| ATTR (Transthyretin) | Transthyretin (TTR) - thyroxine/retinol transport protein | Familial amyloidosis (autosomal dominant); Senile systemic amyloidosis | Mutant TTR is structurally unstable and misfolds; also wild-type TTR can aggregate in elderly |
| Aβ (Beta-amyloid) | β-Amyloid precursor protein (APP) | Alzheimer's disease, Down syndrome | Abnormal cleavage of APP by β- and γ-secretases produces Aβ peptides that aggregate in brain |
| Aβ2M | β2-microglobulin | Hemodialysis-associated amyloidosis | β2-microglobulin (MHC class I component) accumulates because it is not cleared by dialysis membranes |
Why SAA Accumulates in Reactive Amyloidosis
- Chronic inflammation → macrophages and T cells produce IL-6 and IL-1
- These cytokines stimulate hepatocytes to produce large quantities of SAA
- SAA is normally degraded to completion; in some individuals, degradation is incomplete, producing intermediates that are prone to forming fibrils
- Only a subset of patients with chronically elevated SAA develop amyloidosis - suggesting individual variation in SAA catabolism
Net Effect - Tissue Damage
- Compress adjacent parenchymal cells → pressure atrophy
- Obliterate vascular lumens → ischemia
- Replace functional tissue → organ failure (nephrotic syndrome in kidney, restrictive cardiomyopathy in heart, hepatomegaly in liver)
Key exam point: The β-pleated sheet conformation is the structural basis of ALL amyloid, is responsible for Congo red staining and apple-green birefringence under polarized light, and is the reason amyloid resists normal proteolytic degradation.
2 example of red infarction
Two Examples of Red (Hemorrhagic) Infarction
What is a Red Infarct?
"Red infarcts occur: (1) as a result of venous occlusions; (2) in tissues with dual circulations such as the lung and small intestine, where partial, albeit inadequate, perfusion by collateral arterial supplies is typical; (3) in previously congested tissues; and (4) when flow is reestablished after infarction has occurred."
Example 1: Pulmonary Infarction
- Raised, dark red-blue, roughly wedge-shaped (cone-shaped) area of coagulative necrosis
- Base of the wedge is directed toward the pleural surface; apex points toward the hilum (occluded vessel)
- The overlying pleura is often covered by a fibrinous exudate (pleuritis)
Example 2: Testicular Infarction (Testicular Torsion)
- Testis is swollen, dark red to black, firm and hemorrhagic throughout
- Represents complete hemorrhagic infarction
Summary: Why These Two Become Red
| Feature | Pulmonary Infarct | Testicular Infarct |
|---|---|---|
| Mechanism | Dual blood supply - bronchial artery continues to perfuse necrotic tissue | Venous occlusion - blood enters but cannot drain |
| Primary occlusion | Arterial (pulmonary embolism) | Venous (torsion) |
| Shape | Wedge-shaped, pleural base | Diffuse throughout organ |
| Cause | Pulmonary thromboembolism | Testicular torsion |
Other Recognized Causes of Red Infarcts (for completeness):
- Small intestine (dual/collateral arterial supply + venous occlusion in hernia/volvulus)
- Brain - reperfusion injury after arterial recanalization
- Ovarian torsion - venous occlusion (analogous to testis)
- Previously congested organs (e.g., cardiac failure → passive congestion → infarct → hemorrhagic)
Key exam point: Red infarcts = dual circulation OR venous occlusion OR reperfusion. White infarcts = single end-arterial supply (heart, spleen, kidney) with arterial occlusion.