LangerHan cell histocytosis pulmonary
Reading File
Finding Sources
Searching PubMed
"Langerhans cell histiocytosis" AND pulmonary
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive, detailed information from multiple authoritative sources. Let me compile the complete answer.
Pulmonary Langerhans Cell Histiocytosis (PLCH)
Definition & Overview
Pulmonary Langerhans cell histiocytosis (PLCH) is an uncommon diffuse cystic lung disease caused by infiltration and destruction of bronchiolar walls by specialized dendritic cells (Langerhans cells) organized into granulomas. It represents approximately 3-5% of all cases referred for evaluation of interstitial lung disease. It belongs to a broader group called Langerhans cell histiocytosis (LCH) - an umbrella term for proliferative disorders of dendritic cells collectively characterized by clonal proliferation of CD1a+/Langerin+ myeloid cells.
- Murray & Nadel's Textbook of Respiratory Medicine
- Fishman's Pulmonary Diseases and Disorders
Epidemiology
- Age: Peak incidence in the 2nd-4th decade of life; primarily young adults (ages 20-40)
- Sex: Men and women equally affected (females may develop it at slightly older ages)
- Smoking: Strongly smoking-related - >90% of adults with isolated PLCH are active or former smokers
- Cannabis: ~20% of patients in one national registry were also cannabis users
- Race: Occurs mostly in Caucasians; rare in children as an isolated pulmonary form
- Rare familial cases and homozygous twins have been reported
Pathogenesis & Molecular Biology
The pathogenesis involves activation of the MAPK-ERK pathway. The key molecular driver is the BRAF V600E mutation, found in approximately 50% of LCH lesions. This is the same mutation found in hairy cell leukemia, melanoma, papillary thyroid carcinoma, and some colon cancers. BRAF is a component of the RAS signaling pathway, driving proliferation and survival of neoplastic Langerhans cells.
Importantly, gene expression profiling shows LCH cells are more consistent with immature myeloid dendritic cell precursors than classical Langerhans cells of the epidermis. Cigarette smoking is thought to trigger recruitment and abnormal proliferation of these cells in the lung parenchyma.
Robbins & Kumar Basic Pathology, p.424-425
Histopathology
Markers:
- Express MHC class II antigens, CD1a, and Langerin (CD207)
- Birbeck granules on electron microscopy: pentalaminar rod-like tubular structures with characteristic periodicity and sometimes a dilated terminal end giving a "tennis racket" appearance
- Light microscopy: abundant vacuolated cytoplasm and vesicular folded/grooved nuclei (Coffee bean nuclei)
Pathologic stages:
| Stage | Findings |
|---|---|
| Early | Inflammatory bronchiolitis; cellular nodules adjacent to small airways mixed with T lymphocytes, monocytes, eosinophils, plasma cells; CD1a+ cells prominent |
| Intermediate | Bronchiolar destruction; cavitated lesions (residual bronchiolar lumen, NOT necrosis); numbers of CD1a+ cells decrease |
| Advanced | Stellate scars; confluent cystic cavities surrounded by fibrous ring; traction emphysema; predominant bullous/cystic destruction of middle and upper lung zones |
Nodule sizes range 0.1-1.5 cm. Lesions are elongated sheath-like structures extending proximally and distally along bronchioles (3D reconstruction). Vascular infiltration may occur in some cases, explaining pulmonary hypertension as a complication.
Fishman's Pulmonary Diseases and Disorders, p.1039-1040
Imaging
HRCT - The Key Diagnostic Tool
Early disease: Bilateral, symmetric nodules with upper and mid-lung predominance (sparing the costophrenic angles / lung bases). Nodules may be solid or have cavitary centers.
Progression: Mixed pattern of nodules AND thin-walled cysts of varying shapes (round, oval, or bizarre irregular shapes). The coexistence of nodules + cysts in a young smoker is highly suggestive.
Advanced disease: Predominantly large cysts and bullae, often with confluent destruction - can mimic emphysema or lymphangioleiomyomatosis (LAM).
Key distinguishing feature: Upper/mid-lung predominance with relative sparing of costophrenic angles (compare to LAM which is diffuse, and usual interstitial pneumonia which is basal-predominant).
Below is a representative HRCT showing bilateral cysts and nodules in a patient with early-stage PLCH:
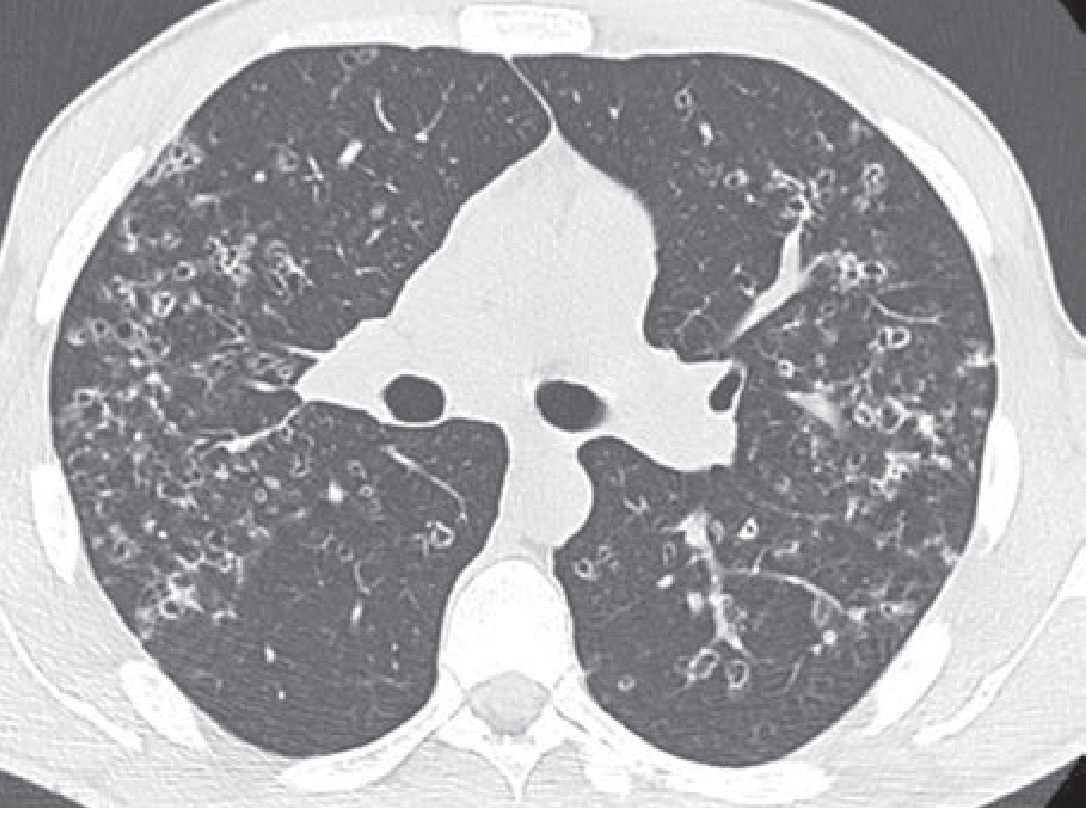
Fishman's Pulmonary Diseases and Disorders - Figure 59-4: Representative HRCT showing characteristic nodules and cysts in early PLCH
Pulmonary Function Tests
| Parameter | Finding |
|---|---|
| DLCO | Reduced in 80-90% of cases (most common abnormality) |
| Vital Capacity | Reduced |
| Residual Volume | Normal or increased (air trapping) |
| Total Lung Capacity | Usually preserved |
| Pattern | Variable - can be obstructive (15-20%), restrictive (~10%), or mixed |
| Early disease | May be normal in 10-15% |
A disproportionately reduced DLCO relative to other lung function parameters suggests pulmonary vascular involvement.
Clinical Features
Symptoms:
- Dry cough and progressive dyspnea (most common)
- Chest pain (pleuritic, from pneumothorax)
- Constitutional symptoms: fever, weight loss, fatigue (especially in multisystem disease)
- ~25% asymptomatic at diagnosis
Complications:
- Pneumothorax - spontaneous, may be recurrent (occurs in ~15-25% of patients)
- Pulmonary hypertension (PH) - a serious complication in advanced disease; classified as Group 5 PH. Severity is often disproportionate to the degree of parenchymal destruction. Mechanisms: vascular remodeling, proliferative vasculopathy including venular involvement (veno-occlusive disease pattern), chronic hypoxemia
- Diabetes insipidus (if hypothalamic-pituitary stalk involved in multisystem LCH)
- Exercise intolerance - VO2 peak reduced in ~72%; multifactorial (ventilatory + cardiovascular + PH)
Diagnosis
Bronchoalveolar lavage (BAL): CD1a+ cells >5% of BAL differential is diagnostic (normal <1%).
Lung biopsy (surgical / transbronchial): Gold standard when imaging is non-diagnostic. Shows cellular nodules adjacent to bronchioles with CD1a+/Langerin+ cells.
Clinical diagnosis can often be made without biopsy in a young smoker with characteristic HRCT findings (upper-lobe-predominant cysts + nodules).
PET scan: Active nodular lesions show FDG uptake; useful for identifying extrapulmonary involvement and assessing disease activity.
Management
1. Smoking cessation - the cornerstone of treatment. Leads to stabilization or improvement in many patients. Abstinence may be the only intervention needed for sustained remission. However, lung function may not always improve after quitting.
2. Monitoring: Longitudinal pulmonary function tests every 3-6 months. Screen for PH with echocardiography (especially with disproportionate dyspnea or declining DLCO).
3. Corticosteroids: Limited evidence; retrospective studies show benefit but confounded by concomitant smoking cessation. Now used infrequently given availability of targeted therapies.
4. Chemotherapy (progressive or multisystem disease):
- Cladribine (2-CdA) - purine nucleoside analog; most evidence; shown to reduce cyst dimensions anecdotally; bone marrow toxicity in young patients is a concern
- Vinblastine + prednisone (traditional for pediatric LCH)
5. Targeted therapy (BRAF V600E mutation present + progressive disease):
- Vemurafenib (BRAF inhibitor) or Trametinib (MEK inhibitor)
- MEK inhibitors may work even in BRAF wild-type progressive disease
- Should be used only at specialized centers given potential toxicity
6. Pulmonary hypertension: Right heart catheterization for confirmation and vasodilator testing; standard PH therapies with caution (vasodilatory drugs may worsen V/Q mismatch)
7. Lung transplantation: For end-stage disease; patients must stop smoking before transplant as disease can recur in the transplanted lung with continued smoking.
Murray & Nadel's Textbook of Respiratory Medicine, pp.2172-2173
Prognosis
- Overall prognosis is good if smoking is stopped early
- Some patients experience spontaneous remission
- ~33% have a progressive course despite treatment
- Median survival is similar to the general population in mild disease
Poor prognostic factors:
- Multisystem involvement
- Extensive cysts on HRCT
- Markedly reduced DLCO
- Low FEV1/FVC ratio
- High RV/TLC ratio
- Pulmonary hypertension
- Young age at presentation
Classification Summary (LCH overall)
| Old Name | Current Name | Features |
|---|---|---|
| Letterer-Siwe disease | Multisystem LCH | Children <2 yrs; skin, liver, spleen, bone, lung; rapidly fatal without treatment |
| Hand-Schüller-Christian disease | Multifocal unisystem LCH | Children; bone lesions + diabetes insipidus + exophthalmos (classic triad) |
| Eosinophilic granuloma | Unifocal/unisystem LCH | Bone (most common); indolent; may heal spontaneously |
| PLCH | Pulmonary LCH | Adults, smoking-related; isolated lung disease |
Recent Evidence
Recent reviews (2024-2025) continue to confirm PLCH as a smoking-related interstitial lung disease with MAPK-ERK pathway mutations:
- A 2025 review (Benattia et al., Clin Chest Med, PMID 41110924) provides updated management, including targeted therapy
- PLCH is increasingly recognized as part of diffuse cystic lung disease requiring distinction from LAM, BHD syndrome, and lymphoid interstitial pneumonia (Franciosi et al., Chest 2025, PMID 39168181)
- Pulmonary hypertension as a complication of rare pulmonary diseases including PLCH is highlighted in a 2025 update (Tagariello et al., Curr Opin Pulm Med 2025, PMID 40575830)
This is a shared conversation. Sign in to Orris to start your own chat.