I have to write a 10 mark answer: A 25 year old male presents with tender xanthomas and a family history of premature cardiovascular disease. His LDL cholesterol levels are markedly elevated. (A) Discuss the metabolism of lipoproteins with emphasis on LDL metabolism (6M) (B) Explain the above biochemical defect and its clinical consequence (2M) (C) Add a note on diagnostic and therapeutic approaches of hyper lipoprotienemia (2M)
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
I now have all the material needed. Let me compose the complete 10-mark answer.
10-Mark Answer: Lipoprotein Metabolism, Familial Hypercholesterolemia, and Hyperlipoproteinemia
(A) Metabolism of Lipoproteins with Emphasis on LDL Metabolism (6 Marks)
Overview of Lipoproteins
Lipoproteins are macromolecular complexes that transport hydrophobic lipids (triacylglycerol, cholesterol esters, phospholipids) through the aqueous plasma. Each particle has a hydrophobic core surrounded by a hydrophilic shell of phospholipids, free cholesterol, and apolipoproteins. The major classes — chylomicrons, VLDL, IDL, LDL, and HDL — are distinguished by density, size, lipid composition, and apolipoprotein content.
1. Exogenous (Dietary) Pathway — Chylomicron Metabolism
Dietary lipids are absorbed in the small intestine, re-assembled into chylomicrons within enterocytes, and secreted into the lymphatics.
- Chylomicrons contain apo B-48, apo C-II, and apo E.
- In the capillaries of adipose and muscle, lipoprotein lipase (LPL) — activated by apo C-II — hydrolyses the core triacylglycerol (TAG), releasing free fatty acids for cellular uptake.
- As >90% of TAG is degraded, the particle shrinks into a chylomicron remnant. The C apolipoproteins (but not apo B-48 or apo E) are returned to HDL.
- Chylomicron remnants are taken up by the liver via receptor-mediated endocytosis mediated by apo E, through the LDL (apo B-100/E) receptor and LDL receptor-related protein-1 (LRP-1). Lysosomal hydrolysis releases amino acids, free cholesterol, and fatty acids; the receptor is recycled.
(Lippincott Illustrated Reviews: Biochemistry, 8e, p. 644)
2. Endogenous Pathway — VLDL and IDL Metabolism
The liver produces VLDL to export endogenous TAG to peripheral tissues.
- Nascent VLDL contains apo B-100 and acquires apo C-II and apo E from circulating HDL.
- LPL in peripheral tissues hydrolyses VLDL TAG, progressively reducing the particle to IDL (intermediate-density lipoprotein).
- IDL may be:
- Taken up directly by the liver via the LDL (apo B-100/E) receptor, or
- Further processed by hepatic lipase → LDL
- Importantly, each LDL particle is derived from a single precursor VLDL particle, as only one molecule of apo B-100 is present and is conserved throughout the cascade. (Harper's Illustrated Biochemistry, 32e, p. 263)
3. LDL Metabolism — Receptor-Mediated Endocytosis ⭐
LDL is the primary cholesterol-carrying lipoprotein in humans, containing ~70% of total plasma cholesterol. Its metabolism is the central focus of this question.
Step-by-step LDL receptor pathway:
| Step | Event |
|---|---|
| [1] | LDL binds to the LDL receptor (a negatively charged glycoprotein) via apo B-100 in clathrin-coated pits on the cell surface |
| [2] | The LDL–receptor complex is internalized by endocytosis as a coated vesicle |
| [3] | The vesicle loses its clathrin coat and fuses with endosomes |
| [4] | Endosomal ATPase lowers pH → LDL dissociates from the receptor; the receptor recycles to the cell surface |
| [5] | LDL is transferred to lysosomes → hydrolysed by lysosomal acid hydrolases → releases free cholesterol, amino acids, fatty acids, and phospholipids |
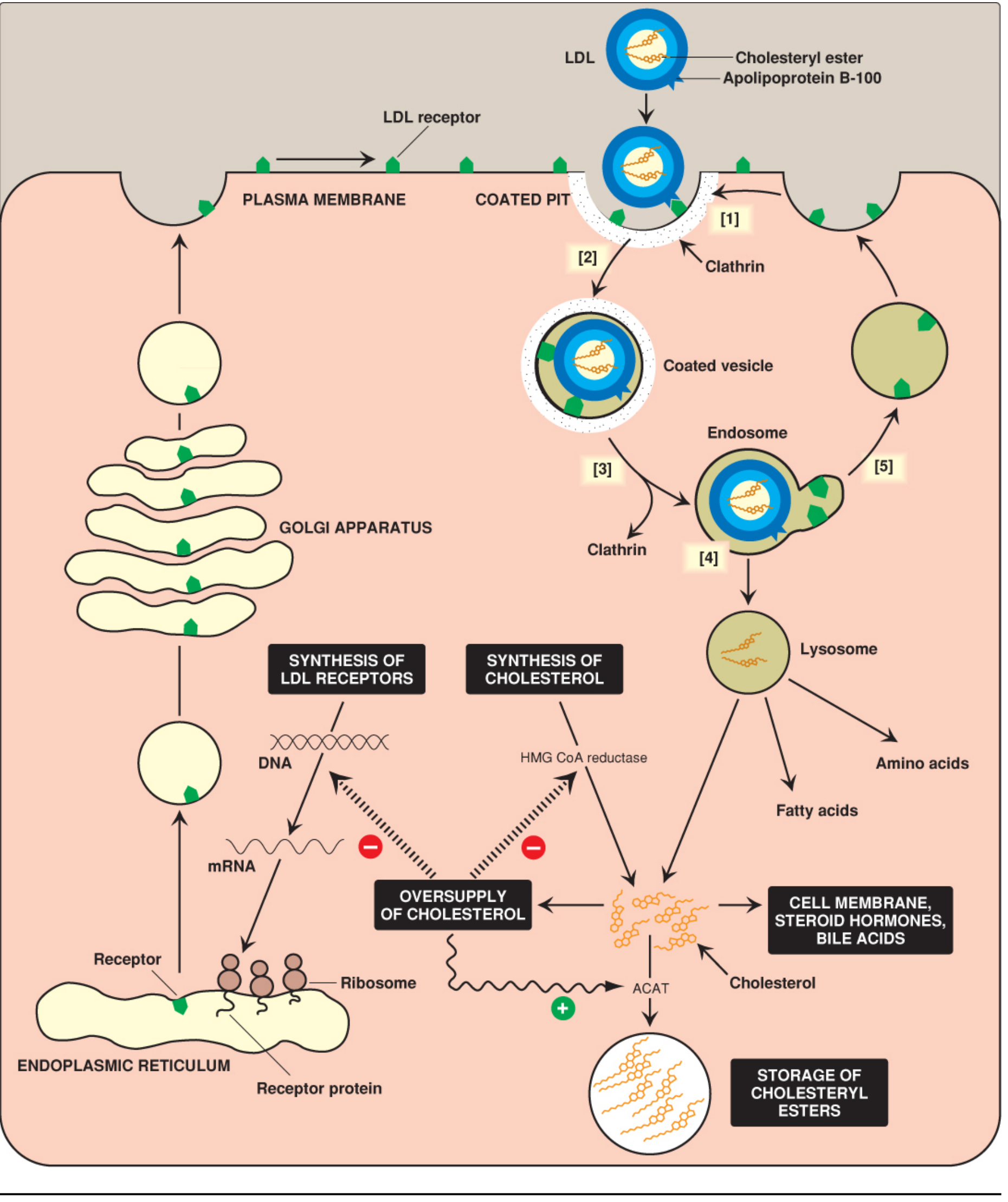
Figure: Cellular uptake and degradation of LDL particles (Lippincott Illustrated Reviews: Biochemistry, 8e, Fig. 18.21)
Cholesterol homeostasis consequences of LDL uptake:
- Suppression of HMG-CoA reductase — excess intracellular cholesterol inhibits transcription of the gene for HMG-CoA reductase (via the SREBP-2 pathway), decreasing de novo cholesterol synthesis; the enzyme is also more rapidly degraded.
- Downregulation of LDL receptors — reduced LDL receptor gene expression via the SRE/SREBP-2 system limits further cholesterol entry, preventing overaccumulation.
- ACAT activation — excess cholesterol is esterified by acyl-CoA:cholesterol acyltransferase (ACAT) and stored as cholesteryl esters in the cytoplasm.
PCSK9 regulation: The protein PCSK9 binds the LDL receptor and targets it for lysosomal degradation rather than recycling, thereby reducing the number of active receptors on the cell surface. (Harper's Illustrated Biochemistry, 32e, p. 275)
Scavenger receptor pathway: Macrophages possess scavenger receptors (SR-A) that bind and internalize oxidised LDL without feedback regulation. This leads to uncontrolled cholesterol ester accumulation, transforming macrophages into foam cells — the hallmark of early atherosclerotic plaque. (Lippincott Illustrated Reviews: Biochemistry, 8e, p. 652)
4. Reverse Cholesterol Transport — HDL Metabolism
HDL acts as a repository of apo C and apo E needed in VLDL/chylomicron metabolism, and performs reverse cholesterol transport (RCT): removing excess cholesterol from peripheral cells → esterification by LCAT → transport to the liver for bile acid synthesis or biliary secretion. This is the basis of HDL's protective role against atherosclerosis.
(B) Biochemical Defect and Clinical Consequences (2 Marks)
Diagnosis: Familial Hypercholesterolemia (FH)
This 25-year-old male with tendinous xanthomas, markedly elevated LDL-C, and a family history of premature cardiovascular disease has classic Familial Hypercholesterolemia (FH) — an autosomal dominant disorder.
Molecular Defect
The most common cause is loss-of-function mutations in the LDL receptor gene (LDLR) on chromosome 19. Other causes include:
- Mutations in apo B-100 (familial defective apoB) — reduce LDL binding affinity to its receptor
- Gain-of-function mutations in PCSK9 — increase receptor degradation, reducing LDL clearance
Heterozygotes (frequency ~1 in 250–500): produce ~50% of normal LDL receptors → plasma LDL-C typically >190 mg/dL.
Homozygotes (~1 in 1 million): virtually no functional LDL receptors → plasma LDL-C >500 mg/dL. (Basic Medical Biochemistry, 6e, p. 1209)
Homozygotes (~1 in 1 million): virtually no functional LDL receptors → plasma LDL-C >500 mg/dL. (Basic Medical Biochemistry, 6e, p. 1209)
Clinical Consequences
| Feature | Mechanism |
|---|---|
| Tendinous xanthomas | Cholesterol deposition in tendons (Achilles, extensor tendons of hands) |
| Xanthelasma | Cholesterol deposits in periorbital skin |
| Corneal arcus | Lipid deposition in corneal stroma (before age 45 = pathological) |
| Premature atherosclerosis | Elevated circulating LDL → oxidative modification → foam cell formation → plaque → coronary artery disease |
| Ischemic heart disease | Significantly increased risk; homozygotes may have MI in childhood |
FH is the most common single-gene cause of premature ASCVD. Heterozygotes are at greatly increased risk of coronary artery disease; homozygotes have severe, often fatal, ischemic heart disease in childhood or early adulthood. (Robbins & Kumar Basic Pathology, p. 151; Harrison's Principles of Internal Medicine, 22e, p. 3286)
(C) Diagnostic and Therapeutic Approaches in Hyperlipoproteinemia (2 Marks)
Diagnosis
1. Lipid Profile (Fasting):
- Total cholesterol, LDL-C, HDL-C, triglycerides
- LDL-C >190 mg/dL in adults should prompt FH evaluation
2. Classification — Fredrickson/WHO Classification:
| Type | Lipoprotein Elevated | Lipid | Example |
|---|---|---|---|
| I | Chylomicrons | ↑↑ TAG | LPL deficiency |
| IIa | LDL | ↑ Cholesterol | Familial hypercholesterolemia |
| IIb | LDL + VLDL | ↑ Cholesterol + TAG | Familial combined hyperlipidemia |
| III | IDL/chylomicron remnants | ↑ Both | Familial dysbetalipoproteinemia (apo E2) |
| IV | VLDL | ↑ TAG | Familial hypertriglyceridemia |
| V | VLDL + Chylomicrons | ↑↑ TAG | Combined |
3. Clinical Criteria (Dutch Lipid Clinic / Simon Broome criteria):
- Family history of premature CVD
- Tendinous xanthomas
- LDL-C levels above age-/sex-specific thresholds
- Genetic testing for LDLR, APOB, PCSK9 mutations confirms diagnosis
4. Additional Tests:
- Coronary artery calcium (CAC) score for risk stratification
- Thyroid function (to exclude secondary causes)
- Liver function, renal function (to exclude secondary hyperlipidemia)
Therapeutic Approaches
Lifestyle Modification (First-line):
- Low saturated fat, high fibre diet
- Weight reduction, aerobic exercise
- Smoking cessation
Pharmacotherapy (Step-wise approach):
| Drug Class | Mechanism | Example |
|---|---|---|
| Statins (first-line) | Inhibit HMG-CoA reductase → ↑ LDL receptor expression → ↑ LDL clearance | Atorvastatin, rosuvastatin |
| Ezetimibe | Inhibits intestinal cholesterol absorption (NPC1L1 transporter) | Used as add-on if statin alone insufficient |
| PCSK9 inhibitors | Monoclonal antibodies that block PCSK9 → prevent LDL receptor degradation → ↑ receptor recycling | Evolocumab, alirocumab |
| Bile acid sequestrants | Bind bile acids in gut → ↑ bile acid synthesis from cholesterol → ↓ hepatic cholesterol | Cholestyramine, colesevelam |
| Bempedoic acid | Inhibits ATP-citrate lyase (upstream of HMG-CoA reductase) | Used in statin-intolerant patients |
Many FH patients require combination therapy — high-intensity statin + ezetimibe ± PCSK9 inhibitor — to achieve LDL-C targets (<70 mg/dL in high-risk patients). Homozygous FH may require LDL apheresis (extracorporeal removal of LDL). (Harrison's Principles of Internal Medicine, 22e, p. 3287)
Summary Table
| Question Part | Key Concept |
|---|---|
| (A) LDL metabolism | Chylomicrons → remnants; VLDL → IDL → LDL; LDL receptor-mediated endocytosis; cholesterol homeostasis via SREBP-2; scavenger receptor/foam cells |
| (B) FH defect | LDLR mutation → impaired LDL uptake → markedly elevated LDL-C → xanthomas + premature atherosclerosis |
| (C) Diagnosis/Rx | Lipid profile, Fredrickson classification, genetic testing; statins → ezetimibe → PCSK9 inhibitors → apheresis |
Sources: Lippincott Illustrated Reviews: Biochemistry, 8e; Harper's Illustrated Biochemistry, 32e; Basic Medical Biochemistry: A Clinical Approach, 6e; Robbins & Kumar Basic Pathology; Harrison's Principles of Internal Medicine, 22e
What are prostaglandins? From which fatty acid are they derived. Explain the role of prostaglandins in humans. Why are they called local hormones? (5M)
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
I now have all the material needed for a comprehensive 5-mark answer. Let me compose it.
Prostaglandins — 5-Mark Answer
Definition
Prostaglandins (PGs) are a family of biologically active, unsaturated 20-carbon fatty acid derivatives that contain an internal cyclopentane (5-carbon) ring, a carboxyl group at C-1, and a hydroxyl group at C-15. They belong to the larger class of compounds called eicosanoids — so named because they all contain 20 carbon atoms (eicosa = 20 in Greek). Along with thromboxanes and prostacyclins, they form the prostanoid sub-family of eicosanoids.
They were first isolated from seminal fluid in the 1930s and were named "prostaglandins" as they were initially believed to originate from the prostate gland. Later work showed they are produced by virtually all nucleated cells in the body.
Precursor Fatty Acid — Arachidonic Acid
The primary precursor is arachidonic acid (AA) — a polyunsaturated ω-6 fatty acid with the structure 20:4, Δ5,8,11,14.
Origin of arachidonic acid:
- Humans cannot synthesise it de novo
- It is derived from the essential fatty acid linoleic acid (18:2, Δ9,12) — an ω-6 fatty acid obtained from dietary plant oils — through a series of elongation and desaturation reactions in the endoplasmic reticulum
- It is stored esterified at the sn-2 position of membrane phospholipids
Three prostaglandin series exist, depending on the precursor:
| Series | Precursor | Double bonds |
|---|---|---|
| 1-series (PG₁) | Dihomo-γ-linolenic acid (20:3) | 1 (Δ13,14) |
| 2-series (PG₂) | Arachidonic acid (20:4) | 2 (Δ5,6 + Δ13,14) — major human series |
| 3-series (PG₃) | Eicosapentaenoic acid/EPA (20:5) | 3 |
(Basic Medical Biochemistry: A Clinical Approach, 6e, p. 1136)
Synthesis — The Cyclooxygenase Pathway
Step 1 — Release of arachidonic acid:
Stimuli (hormones, cytokines, tissue injury, histamine) activate phospholipase A₂ (or phospholipase C), which cleaves arachidonic acid from membrane phospholipids.
Step 2 — Cyclooxygenase (COX) pathway:
Free arachidonic acid is metabolised by cyclooxygenase (prostaglandin H synthase), which has two enzymatic activities:
- COX activity: converts AA → PGG₂ (with addition of two O₂)
- Peroxidase activity: reduces PGG₂ → PGH₂ (the common intermediate)
PGH₂ is then converted by tissue-specific enzymes into the various prostaglandins (PGE₂, PGD₂, PGF₂α, PGI₂/prostacyclin) and thromboxane A₂ (TXA₂).
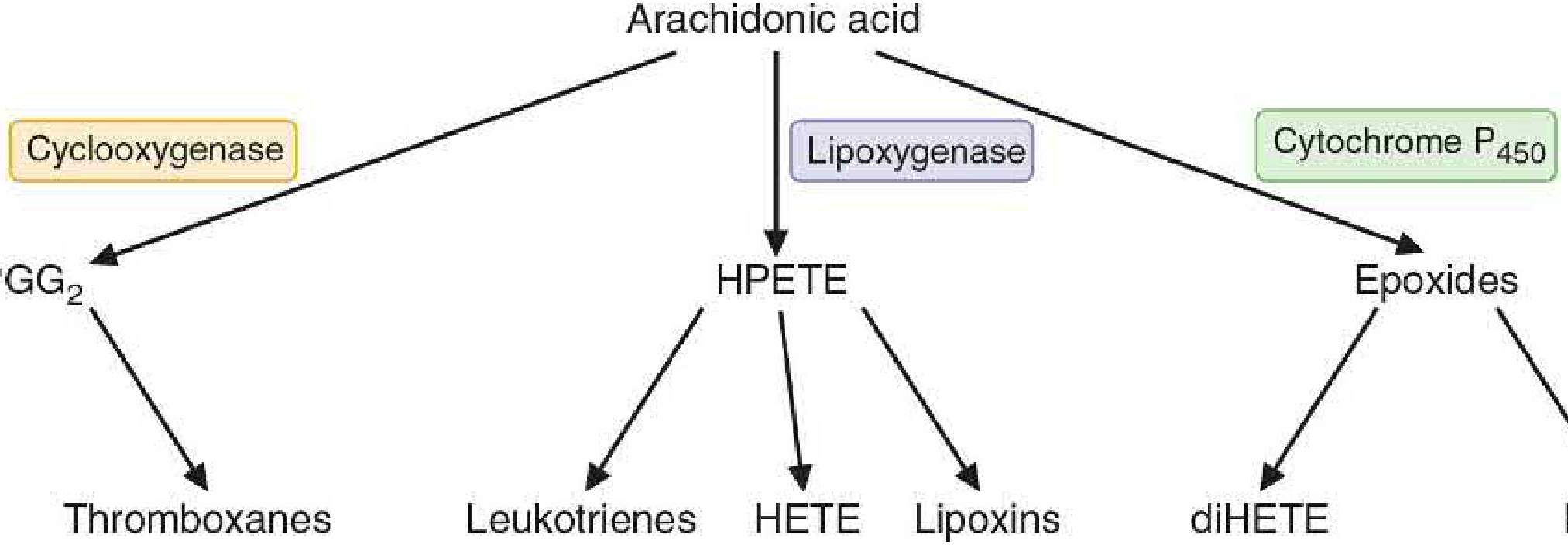
Figure: Three metabolic pathways of arachidonic acid (Basic Medical Biochemistry: A Clinical Approach, 6e, Fig. 31.16)
Two isoforms of cyclooxygenase:
- COX-1 — constitutive; present in nearly all tissues including platelets; mediates physiological ("housekeeping") prostaglandin production (gastric protection, platelet function, renal blood flow)
- COX-2 — inducible; expressed at low levels normally but markedly upregulated at sites of inflammation by cytokines (TNF-α, IL-1) and oxidative stress; inhibited by glucocorticoids
(Lippincott Illustrated Reviews: Pharmacology, p. 1334–1335)
Why Are They Called "Local Hormones"?
Prostaglandins are called local hormones (or autocoids / paracrine mediators) for several distinct reasons:
-
Ubiquitous synthesis — They are produced in minute quantities by virtually all tissues (unlike classical hormones which are made by discrete endocrine glands), wherever stimuli are applied.
-
Local action — They act on or near the cells that produce them (autocrine and paracrine action), not on distant target organs via the bloodstream as classical hormones do. They exert effects at the site of synthesis.
-
Rapid inactivation — They are rapidly metabolised to inactive products at their site of action and do not circulate in the blood in significant concentrations. Half-life in plasma is only seconds to minutes. For example, PGI₂ (prostacyclin) has a half-life of ~2 minutes. This rapid degradation prevents systemic hormonal-like actions.
-
Receptor-mediated mechanism — Like hormones, they act through specific G-protein coupled receptors (GPCRs) on target cell membranes, triggering intracellular second messenger cascades (↑cAMP or ↑IP₃/DAG).
-
Potency at very low concentrations — They exert powerful effects at nanomolar or picomolar concentrations, a hallmark of hormone-like signalling.
"Prostaglandins and related compounds are produced in minute quantities by virtually all tissues. They generally act locally on the tissues in which they are synthesized and are rapidly metabolized to inactive products at their sites of action. Therefore, prostaglandins do not circulate in the blood in significant concentrations." — Lippincott Illustrated Reviews: Pharmacology, p. 1333
Physiological and Pathological Roles of Prostaglandins
| Prostaglandin | Tissue | Major Action |
|---|---|---|
| PGI₂ (prostacyclin) | Vascular endothelium | Vasodilation; ↓ platelet aggregation; protects vessel wall |
| TXA₂ | Platelets | Vasoconstriction; ↑ platelet aggregation (antagonises PGI₂) |
| PGE₂ | Stomach | ↑ Mucus & bicarbonate secretion; ↓ gastric acid — cytoprotection |
| PGE₂, PGI₂ | Hypothalamus | Fever (pyrogen action via ↑ cAMP in thermoregulatory centre) |
| PGE₂, PGI₂ | Peripheral sensory neurons | Sensitise nociceptors → pain and hyperalgesia |
| PGE₂, PGD₂ | Bronchial smooth muscle | Bronchodilation |
| PGF₂α | Uterus / bronchi | Uterine contraction; bronchoconstriction |
| PGE₂ | Kidney | Maintain renal blood flow; regulate sodium excretion; renin release |
| PGD₂ | Mast cells / brain | Allergic responses; sleep regulation |
| PGE₂, PGI₂ | Blood vessels | Vasodilation; ↓ leukocyte aggregation; modulate inflammation |
Key roles summarised:
- Inflammation: PGs (especially PGE₂) are cardinal mediators of the four signs — they promote vasodilation (redness, warmth), increase vascular permeability (swelling), and sensitise pain receptors (pain). COX-2-derived PGs dominate in inflammation.
- Fever: Bacterial pyrogens → macrophage IL-1 → hypothalamic COX-2 → ↑ PGE₂ → reset thermostat → fever. NSAIDs and aspirin reduce fever by blocking COX.
- Haemostasis: The TXA₂/PGI₂ balance determines platelet aggregation and vascular tone — a critical check in haemostasis.
- Reproduction: PGF₂α stimulates uterine contraction; used clinically to induce labour and terminate pregnancy (e.g., dinoprostone/misoprostol).
- GI protection: PGE₁ and PGE₂ maintain the gastric mucosal barrier. NSAID-induced gastric ulcers result from inhibiting this protective prostaglandin synthesis.
- Renal function: PGs help maintain renal cortical blood flow during states of reduced perfusion; NSAIDs can precipitate acute kidney injury in at-risk patients.
Summary Diagram
Membrane phospholipids
↓ (Phospholipase A₂ — activated by stimuli)
Arachidonic acid (20:4, ω-6)
↓ (COX-1 / COX-2)
PGG₂ → PGH₂
↙ ↓ ↓ ↘
PGE₂ PGD₂ PGF₂α PGI₂ TXA₂
Sources: Basic Medical Biochemistry: A Clinical Approach, 6e; Lippincott Illustrated Reviews: Pharmacology; Robbins & Cotran Pathologic Basis of Disease
This is a shared conversation. Sign in to Orris to start your own chat.