Recurrent hypoglycaemia in newborn
Reading File
Finding Sources
Finding Sources
Searching PubMed
"neonatal hypoglycemia" AND "recurrent" AND management
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
I now have comprehensive information to write a thorough answer.
Recurrent Hypoglycaemia in the Newborn
Definition
Neonatal hypoglycaemia is defined as a plasma glucose insufficient to meet metabolic requirements. Practically:
- <45-50 mg/dL (point-of-care) within the first 48 hours of life
- <70 mg/dL beyond 48 hours of life
Note: Bedside glucometers can be inaccurate by 10-15 mg/dL in the hypoglycaemic range - a STAT plasma glucose must always confirm the diagnosis.
Recurrent or persistent hypoglycaemia (beyond 48-72 hours, or requiring high glucose infusion rates >8 mg/kg/min) demands a structured diagnostic workup for an underlying cause.
Pathophysiology of Glucose Homeostasis in the Newborn
The normal newborn is highly dependent on hepatic glycogen stores during the first day of life. Mature fasting systems (gluconeogenesis, ketogenesis) develop after the first 24 hours. Plasma glucose falls below 50 mg/dL in approximately one-third of normal infants during the first 6 hours - by day 2, this frequency falls to <0.5%. This physiological nadir resolves spontaneously with feeding.
Recurrent hypoglycaemia reflects failure of normal counter-regulatory mechanisms: inadequate glucagon surge, catecholamine deficiency, or - most commonly beyond 7 days - inappropriate insulin excess.
Causes of Recurrent / Persistent Neonatal Hypoglycaemia
1. Hyperinsulinism (Most Common Cause Beyond Day 7)
Hyperinsulinism is the most common cause of neonatal hypoglycaemia beyond the first 7 days of life. It suppresses both ketogenesis and lipolysis, depriving the brain of alternate fuels - this makes it particularly neurotoxic.
A. Transient Hyperinsulinism
| Setting | Features |
|---|---|
| Infant of a diabetic mother (IDM) | Large/plethoric infant; resolves in 1-2 days |
| Perinatal stress (asphyxia, IUGR, prematurity, maternal toxaemia) | Persists weeks-months; median resolution 6 months |
| Erythroblastosis fetalis | Associated with islet cell hyperplasia |
- IDM hypoglycaemia is driven by fetal hyperinsulinism from prolonged intrauterine hyperglycaemia; it usually resolves within 1-2 days with feeds. If it persists, further evaluation is mandatory.
- Perinatal stress-induced hyperinsulinism: neonates have high glucose utilisation, inappropriately normal or elevated insulin at the time of hypoglycaemia, suppressed beta-hydroxybutyrate, and a glucagon-responsive glucose rise of ~30 mg/dL. Responds well to diazoxide.
B. Congenital Hyperinsulinism (CHI)
- Incidence: 1/50,000 live births (up to 1/2,500 in consanguineous populations)
- Infants are typically large-for-gestational-age, with severe hypoglycaemia requiring glucose infusion rates >10 mg/kg/min
Genetic mechanisms (see diagram below):
Glucose enters the beta cell via GLUT2, is phosphorylated by glucokinase (GCK), raising the ATP/ADP ratio. This closes the KATP channel (SUR1 + Kir6.2 subunits), depolarises the membrane, opens voltage-gated Ca²+ channels, and triggers insulin release.
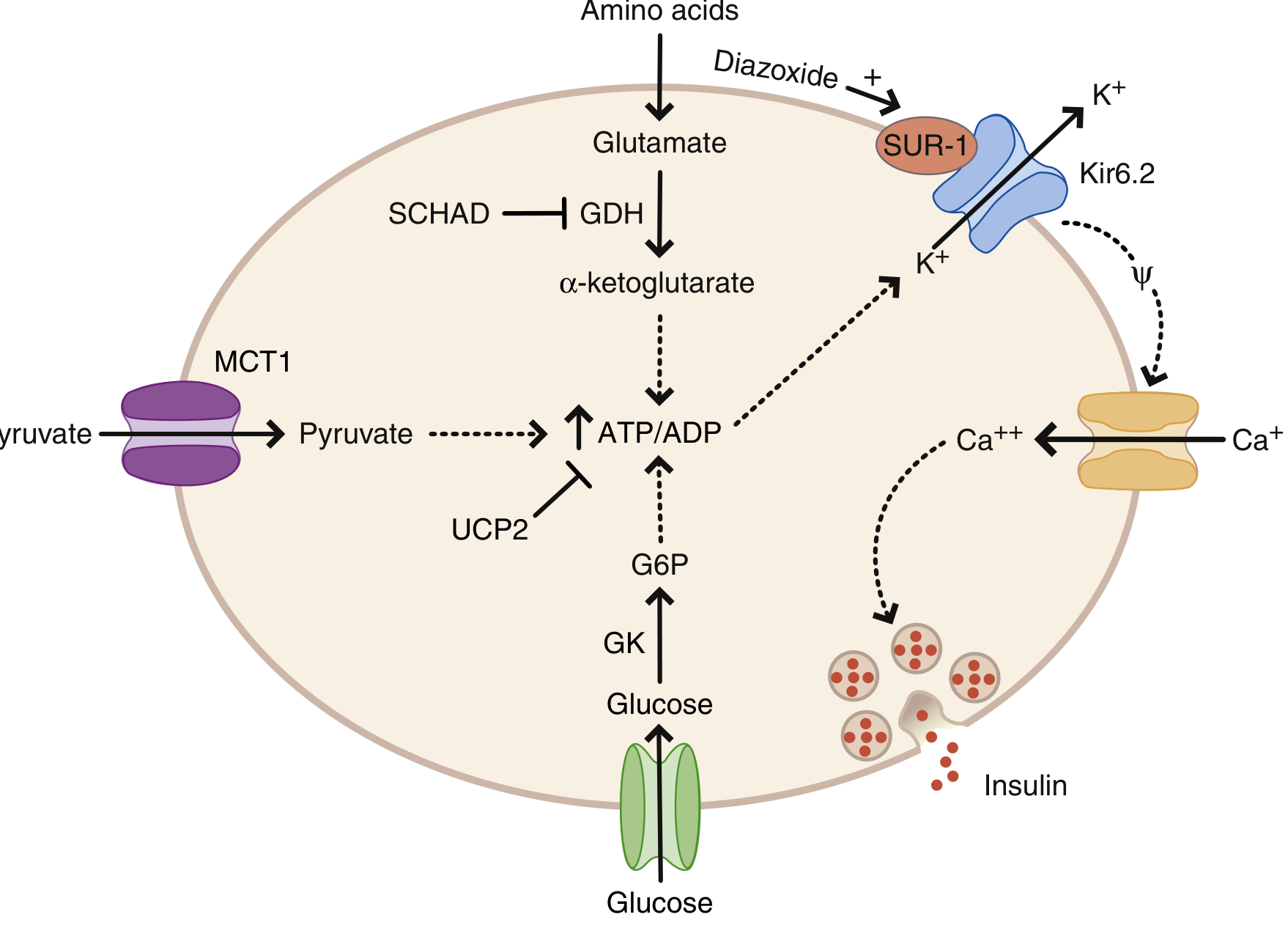
| Gene | Mechanism | Diazoxide response |
|---|---|---|
| ABCC8 (SUR1) | Loss-of-function (KATP closed) | Usually NO |
| KCNJ11 (Kir6.2) | Loss-of-function (KATP closed) | Usually NO |
| GCK (glucokinase) | Gain-of-function (increased ATP) | NO |
| GLUD1 (glutamate dehydrogenase) | Gain-of-function - also causes hyperammonaemia | YES |
| SCHAD (HADH) | Loss-of-function - removes GDH inhibition | YES |
| HNF4A, HNF1A | Transcription factor mutations | YES |
- KATP mutations are the most common and most severe form; most require pancreatectomy
- Focal vs. diffuse CHI can be distinguished by 18F-DOPA PET scan - focal disease may be cured by limited pancreatectomy
2. Counter-regulatory Hormone Deficiencies
Hypopituitarism / GH + ACTH deficiency
- Incidence of hypoglycaemia in hypopituitarism: ~20%
- Key clinical clues: midline defects (septo-optic dysplasia, cleft palate), micropenis (gonadotropin deficiency), cholestatic jaundice
- In the neonatal period, beta-hydroxybutyrate is NOT elevated (unlike older children)
- Low GH and cortisol at the time of hypoglycaemia confirm the diagnosis
Isolated GH deficiency or cortisol deficiency (CAH, Addison)
- CAH (21-hydroxylase): hypoglycaemia + salt-wasting + ambiguous genitalia
3. Inborn Errors of Metabolism
| Disorder | Key Features |
|---|---|
| Fatty acid oxidation defects (e.g. MCAD) | Hypoketotic hypoglycaemia, dicarboxylic aciduria |
| Glycogen storage diseases (GSD I - Von Gierke) | Fasting hypoglycaemia, hepatomegaly, lactic acidosis |
| Galactosaemia | Hypoglycaemia + liver disease after lactose feeds |
| Hereditary fructose intolerance | After introduction of fructose/sucrose |
| Organic acidaemias | Metabolic acidosis, elevated lactate/ammonia |
| Amino acid disorders (maple syrup urine disease) | Encephalopathy, urine odour |
4. Other Causes
- Beckwith-Wiedemann syndrome (macroglossia, omphalocele, macrosomia, ear creases) - due to islet cell hyperplasia/CHI
- Kabuki syndrome - CHI documented
- Costello syndrome (HRAS mutations) - recurrent hypoglycaemia
- SGA/IUGR - depleted glycogen stores + hormonal immaturity
Diagnostic Approach to Recurrent Neonatal Hypoglycaemia
"Critical sample" at the time of hypoglycaemia (plasma glucose <45-50 mg/dL)
| Sample | Interpretation |
|---|---|
| STAT plasma glucose | Confirm hypoglycaemia |
| Serum insulin | Detectable insulin during hypoglycaemia = hyperinsulinism |
| C-peptide | Distinguishes endogenous vs. exogenous insulin |
| Beta-hydroxybutyrate | <2.0 mmol/L = suppressed ketogenesis (hyperinsulinism or FA oxidation defect) |
| Free fatty acids | <1.5 mmol/L = suppressed lipolysis (hyperinsulinism) |
| Cortisol | Low = adrenal insufficiency / hypopituitarism |
| Growth hormone | Low = GH deficiency |
| Ammonia | Elevated in GDH mutation (GLUD1) |
| Lactate | Elevated in GSD, organic acidaemias |
| Urine ketones + organic acids | Metabolic disorders |
| Acylcarnitine profile | Fatty acid oxidation defects |
Glucagon stimulation test
Administer glucagon 1 mg IV/IM; measure glucose Q10 min x4.
- Rise ≥30 mg/dL = glycogenolytic response intact, suggests hyperinsulinism (glycogen stores full due to high insulin)
- Blunted rise = glycogen depletion, GSD, or gluconeogenic defect
Glucose infusion rate (GIR)
- Normal newborn: 4-6 mg/kg/min
- >8 mg/kg/min needed to maintain normoglycaemia = strong indicator of hyperinsulinism
- >10 mg/kg/min = congenital hyperinsulinism until proven otherwise
Profile interpretation
| Pattern | Likely Diagnosis |
|---|---|
| Detectable insulin, low BHB, low FFA, GIR >8, glucagon-responsive | Hyperinsulinism |
| Low insulin, low BHB, low FFA | Fatty acid oxidation defect |
| Low insulin, HIGH BHB, HIGH FFA | GH/cortisol deficiency, ketotic hypoglycaemia |
| Metabolic acidosis + elevated lactate | GSD type I, organic acidaemia |
| Elevated ammonia | GDH mutation (GLUD1) |
Management
Immediate (any neonatal hypoglycaemia)
- Enteral feeds (breast or formula) - first-line if clinically stable
- Buccal dextrose gel (40%) - 200 mg/kg; effective in at-risk term neonates
- IV dextrose - 10% dextrose, 2 mL/kg bolus (200 mg/kg), then continuous infusion titrated to maintain glucose
Treatment Goals
- Suspected congenital hypoglycaemia disorder: maintain plasma glucose >70 mg/dL
- High-risk neonate, no congenital disorder: >45-50 mg/dL at <48 hours; >60 mg/dL at >48 hours
For Persistent / Recurrent Hypoglycaemia (Hyperinsulinism)
Diazoxide - first-line medical therapy
- Mechanism: opens beta-cell ATP-sensitive K+ channels (KATP), hyperpolarises membrane, inhibits insulin release
- Dose: 5-15 mg/kg/day in 2-3 divided doses
- Requires concurrent chlorothiazide to counteract fluid retention
- Black box warning: rarely associated with pulmonary hypertension in neonates (requires echocardiogram before and during use)
- Effective in: perinatal stress hyperinsulinism, GDH/GLUD1, SCHAD, HNF4A/1A mutations
- NOT effective in: KATP channel mutations (ABCC8/KCNJ11)
Octreotide (somatostatin analogue)
- Second-line; inhibits insulin secretion via Gi-protein
- Used when diazoxide fails or is contraindicated
- Risk of necrotising enterocolitis, tachyphylaxis
Glucagon infusion
- Useful as a bridge while establishing diagnosis or preparing for surgery
- 0.01-0.02 mg/kg/hour IV
Surgery (pancreatectomy)
- For diazoxide-unresponsive CHI
- Focal lesion (KATP mutation): localised resection after 18F-DOPA PET - potentially curative
- Diffuse disease: near-total pancreatectomy (>95-98%) - risk of diabetes and exocrine insufficiency
For Counter-regulatory Deficiencies
- GH deficiency: recombinant GH
- Cortisol deficiency / CAH: hydrocortisone replacement
For Metabolic Disorders
- Disease-specific dietary management (e.g. avoid fasting in MCAD, restrict galactose in galactosaemia)
Neurodevelopmental Consequences
Recurrent or prolonged hypoglycaemia carries serious risk of brain injury. Studies show that neonates hypoglycaemic for >2 hours have significantly lower neurodevelopmental adaptability scores at age 2 years compared to controls. Untreated or under-treated hypoglycaemia can lead to seizures, coma, and permanent brain damage. Posterior white matter and occipital cortex are particularly vulnerable.
The target plasma glucose for neonates with confirmed or suspected congenital hypoglycaemia disorders is >70 mg/dL to protect the developing brain.
Summary Algorithm
Recurrent/Persistent Neonatal Hypoglycaemia
|
Obtain Critical Sample at nadir
|
┌───────┴────────┐
Insulin Insulin
detectable undetectable
| |
Hyperinsulinism ┌──┴──────────────┐
| Low BHB/FFA High BHB/FFA
GIR >8? (FA Oxidation) (GH/Cortisol
| Defect) deficiency)
Diazoxide
trial
|
Responsive? → Yes: Perinatal stress / GDH / HNF
No? → Genetic testing + 18F-DOPA PET
→ Surgery if focal CHI
Sources:
- The Harriet Lane Handbook (23rd ed.), Chapter 10 - Neonatal Hypoglycaemia Evaluation
- Henry's Clinical Diagnosis and Management by Laboratory Methods - Hypoglycaemia in Infancy and Childhood (Congenital Hyperinsulinism)
- Creasy & Resnik's Maternal-Fetal Medicine - Neonatal Hypoglycaemia
- Recent reviews: Garg & Devaskar, Eur J Pediatr 2025 (PMID 40119223) on long-term impacts and safe glucose thresholds
Serziurs in new newborn
Reading File
Finding Sources
Finding Sources
Searching PubMed
"neonatal seizures" AND management
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading Article
PMID: 37655702
I now have very comprehensive information. Let me compile the full, detailed answer.
Seizures in the Newborn (Neonatal Seizures)
Neonatal seizures are the most common neurological emergency in newborns and occur more frequently in the first 28 days of life than at any other time. They represent one of the most important manifestations of neonatal brain injury and demand prompt recognition, evaluation, and treatment.
Why Neonatal Seizures Are Different
The neonatal brain is fundamentally different from the adult brain in two ways that directly affect seizure presentation and treatment:
1. Clinical presentation is often subtle
Unlike older children or adults, neonates rarely have classic generalized tonic-clonic seizures. Because the neonatal cortex is immature and lacks complete corticocortical myelination, seizures cannot spread bihemispherically in the same way. Up to 50% of neonatal seizures are subtle, presenting as:
- Lip-smacking or chewing
- Eye deviation or blinking
- Bicycling or pedalling leg movements
- Apnoeic episodes with colour change (pallor/cyanosis)
- Tonic posturing of a limb
- Stiffening of the body
These can be easily confused with normal neonatal jitteriness or myoclonus.
2. GABA is excitatory, not inhibitory, in the immature brain
In adult neurons, GABA-A receptor activation causes chloride influx → membrane hyperpolarisation → inhibition. In immature neurons, the balance of chloride transporters is reversed:
- NKCC1 (Na-K-Cl cotransporter 1) drives Cl- into the cell, keeping intracellular Cl- high
- KCC2 (K-Cl cotransporter 2) - which extrudes Cl- - is underexpressed at birth
The result: GABA-A activation in the neonatal brain causes Cl- efflux → membrane depolarisation → excitation. This is a key reason why phenobarbital (a GABA-A agonist) often has a disappointing response in neonates.
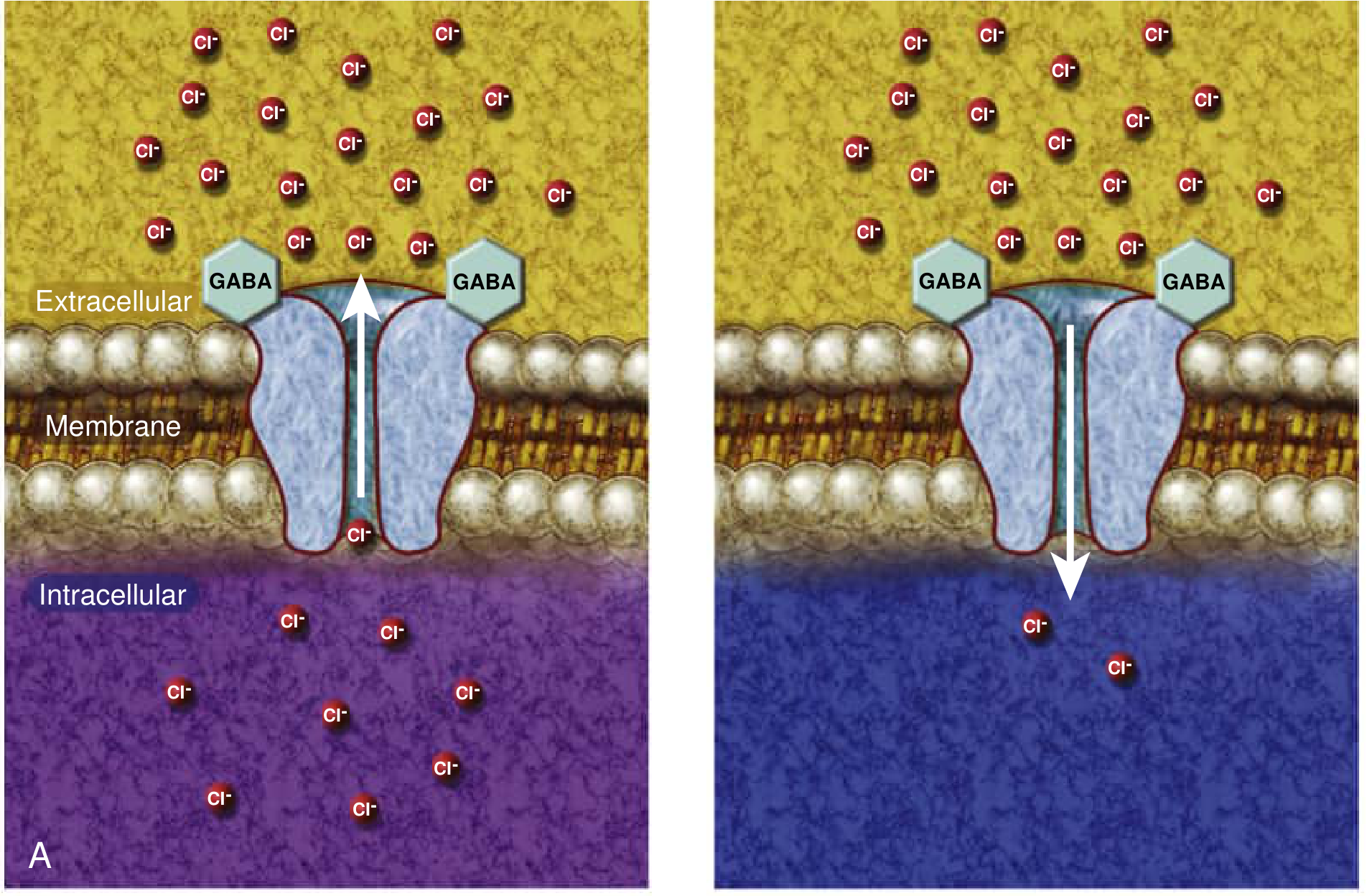
Etiology - By Day of Presentation
The timing of seizure onset is the most important clue to aetiology:
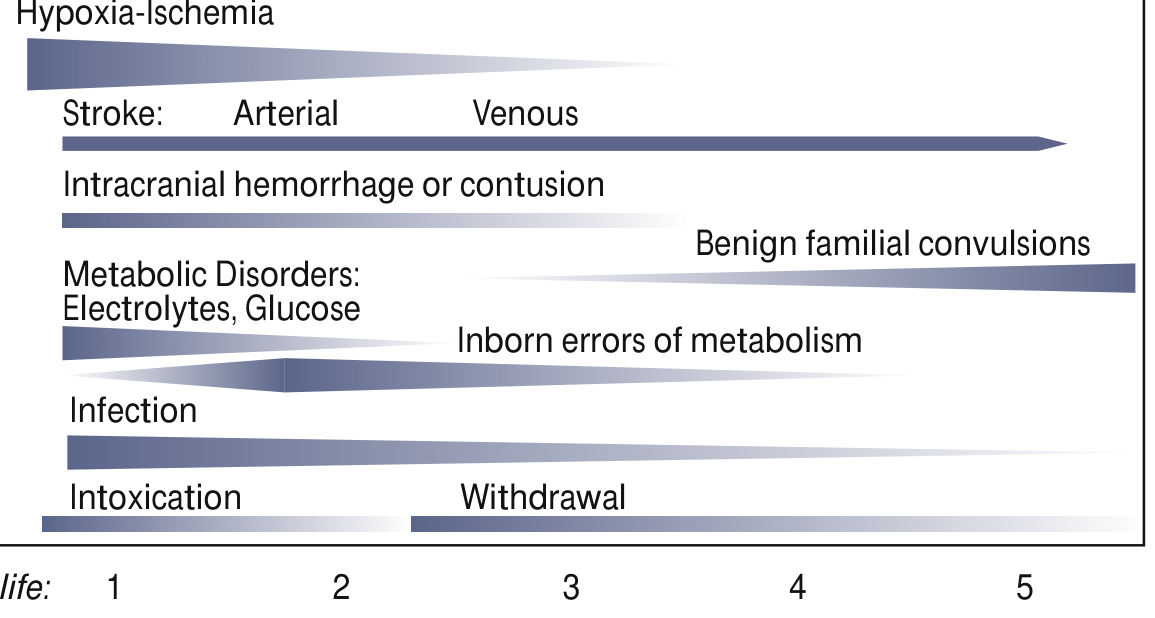
Day 1 (Most Common Causes)
| Cause | Key Features |
|---|---|
| Hypoxic-Ischaemic Encephalopathy (HIE) | Most common cause (~50% in term infants); difficult delivery, low Apgar scores, metabolic acidosis; seizures within 24-48h of birth |
| Hypoglycaemia | Blood glucose <45 mg/dL; especially IDM (infant of diabetic mother), SGA |
| Electrolyte disturbances | Hypocalcaemia (<7 mg/dL), hypomagnesaemia, hyponatraemia |
| Intracranial haemorrhage | IVH in premature; subdural/subarachnoid in term |
| Intoxication / withdrawal | Maternal narcotics, cocaine, SSRI |
Days 2-3
| Cause | Key Features |
|---|---|
| Perinatal stroke (arterial/venous) | Focal seizures; often unilateral clonic activity |
| Metabolic disorders | Electrolyte abnormalities peak |
| Infection | Bacterial meningitis (Group B Strep, E. coli, Listeria), HSV encephalitis |
Days 3-7+
| Cause | Key Features |
|---|---|
| Inborn errors of metabolism | Pyridoxine deficiency, non-ketotic hyperglycinaemia, maple syrup urine disease, organic acidaemias |
| Benign familial neonatal seizures | KCNQ2/KCNQ3 mutations; onset day 2-3, remit by 1 year |
| Congenital brain malformations | Cortical dysplasia, lissencephaly, holoprosencephaly |
| Drug withdrawal | Peaks days 2-4 (opioids) |
| Infection | HSV, TORCH infections (CMV, toxoplasma, rubella) |
Special note on prematurity: In preterm newborns, HIE and intracranial haemorrhage each account for approximately one-third of seizures.
Seizure Types (Neonatal Classification)
| Type | Description |
|---|---|
| Subtle | Most common; eye deviation, lip-smacking, apnoea, bicycling |
| Focal clonic | Rhythmic jerking of one limb; well localised; often perinatal stroke |
| Focal tonic | Sustained posturing of one limb |
| Multifocal clonic | Sequential jerking of multiple limbs; metabolic or HIE |
| Myoclonic | Brief, rapid jerks; metabolic encephalopathy; worse prognosis |
| Generalised tonic | Uncommon; associated with severe injury (IVH, hypoxia) |
Electroclinical dissociation is important: in neonates, clinical seizure activity may occur without EEG changes (motor automatisms) and - critically - EEG seizures may occur without any clinical manifestation (subclinical/electrographic-only seizures).
Neonatal Epilepsy Syndromes
Benign
- Benign familial neonatal epilepsy (BFNE): autosomal dominant mutations in KCNQ2, KCNQ3, or SCN2A (voltage-gated K+/Na+ channels); onset first week; remit within first year; good neurodevelopment
- "Fifth-day fits": non-familial; onset days 4-6; partial seizures, discontinuous EEG theta; good prognosis
Severe (Epileptic Encephalopathies)
- Ohtahara syndrome / Early Infantile Epileptic Encephalopathy (EIEE): intractable tonic seizures + burst-suppression EEG; genetic aetiology; may evolve into West syndrome
- Early Myoclonic Encephalopathy (EME): erratic shifting myoclonus + burst-suppression; often metabolic (non-ketotic hyperglycinaemia)
- KCNQ2 encephalopathy: neonatal seizures with EEG burst-suppression; responds to sodium channel blockers
Treatable Metabolic Epileptic Encephalopathies (Must Not Miss)
These are rare but reversible causes that must be considered in seizures refractory to standard drugs:
| Disorder | Treatment |
|---|---|
| Pyridoxine (B6) dependency (ALDH7A1/antiquitin deficiency) | Pyridoxine 100 mg IV - response within minutes |
| Pyridoxal-5-phosphate (PLP) responsive epilepsy (PNPO deficiency) | Pyridoxal-5-phosphate (NOT pyridoxine) |
| Folinic acid-responsive seizures (allelic to ALDH7A1) | Folinic acid |
| Biotinidase deficiency | Biotin |
| Non-ketotic hyperglycinaemia (NKH) | Sodium benzoate, dextromethorphan |
| GLUT1 deficiency | Ketogenic diet |
| 3-phosphoglycerate dehydrogenase deficiency | Serine + glycine |
Diagnosis
Clinical Assessment
- Full birth history (asphyxia, prematurity, instrumental delivery)
- Maternal history (diabetes, drug use, infection, medications - beta-blockers)
- Family history (genetic epilepsy syndromes)
- Physical exam: dysmorphic features (genetic syndrome), anterior fontanelle (raised ICP), skin lesions (HSV, tuberous sclerosis), microcephaly
Immediate Investigations (Emergency)
| Test | Rationale |
|---|---|
| Bedside blood glucose | Hypoglycaemia - first priority |
| Blood gases | Acidosis in HIE, metabolic disorders |
| Electrolytes: Na, Ca, Mg | Hyponatraemia, hypocalcaemia, hypomagnesaemia |
| FBC | Infection, polycythaemia |
| Blood culture, LP (CSF) + PCR for HSV | Meningitis/encephalitis |
| CRP, procalcitonin | Infection screen |
Second-Line Investigations
| Test | Rationale |
|---|---|
| Cranial ultrasound | IVH (especially preterm), periventricular leukomalacia |
| MRI brain (preferred) | HIE, stroke, malformation, cortical dysplasia |
| Head CT | If non-accidental trauma or haemorrhage suspected |
| Serum lactate, ammonia | Metabolic disorders |
| Serum amino acids, urine organic acids | Inborn errors of metabolism |
| Toxicology screen | Maternal drug exposure |
| Acylcarnitine profile | Fatty acid oxidation disorders |
EEG - the Gold Standard
- Continuous video-EEG monitoring is the definitive tool to detect and quantify seizures - especially electrographic-only seizures
- Amplitude-integrated EEG (aEEG): bedside tool; useful for monitoring but may miss focal seizures
- Neonatal EEG findings include discontinuous theta activity, focal sharp waves, and burst-suppression patterns
Management
Step 1: Treat the Cause First
Always correct reversible causes before or alongside anti-seizure medications (ASMs):
- Hypoglycaemia: 10% dextrose 2 mL/kg IV bolus
- Hypocalcaemia: Calcium gluconate 10% - 1-2 mL/kg IV slowly
- Hypomagnesaemia: Magnesium sulphate 50% - 0.2 mL/kg IV/IM
- Infection: Empiric IV antibiotics (ampicillin + gentamicin) + acyclovir (for HSV)
- Pyridoxine trial: 100 mg IV if seizures refractory to 2nd-line agents, with EEG monitoring
Step 2: Anti-Seizure Medications (ASMs)
Based on the 2023 ILAE Task Force Guidelines (PMID 37655702):
First-Line: Phenobarbital
- Dose: 20 mg/kg IV loading dose; may repeat 10 mg/kg x1-2 if seizures persist (max 40 mg/kg)
- Maintenance: 3-5 mg/kg/day in 1-2 divided doses
- Effective in ~50% of neonatal seizures
- Mechanism: enhances GABA-A, inhibits glutamate
- Limitation: GABA is excitatory in immature neurons (reduced efficacy); respiratory depression risk
- Exception: If channelopathy suspected (family history of genetic epilepsy), use phenytoin or carbamazepine (sodium channel blockers) as first line
Second-Line Options (if phenobarbital fails)
| Drug | Dose | Notes |
|---|---|---|
| Phenytoin / fosphenytoin | 20 mg/kg IV load | Effective for channelopathies; cardiac monitoring required |
| Levetiracetam | 40-60 mg/kg IV load | Preferred 2nd-line if cardiac disorder; good safety profile |
| Midazolam | 0.15 mg/kg IV bolus, then infusion 0.1-0.4 mg/kg/h | Benzodiazepine; respiratory monitoring needed |
| Lidocaine | 2 mg/kg IV bolus, then infusion | Effective; NOT if phenytoin already given (cardiac risk) |
Pyridoxine (Vitamin B6)
- 100 mg IV with EEG monitoring - seizure cessation within minutes supports pyridoxine-dependent epilepsy
- A trial should be attempted in any neonate with seizures unresponsive to second-line ASMs
Step 3: When to Stop ASMs
The ILAE 2023 guidelines recommend:
- After acute provoked seizures resolve and there is no evidence of neonatal-onset epilepsy - stop ASMs before discharge, regardless of MRI or EEG findings
- This applies even if MRI shows brain injury - maintenance ASMs do not improve outcomes in this context
Therapeutic Hypothermia
- In HIE, therapeutic hypothermia (33-34°C for 72 hours) reduces seizure burden and improves neurodevelopmental outcome
- An evidence-based ILAE recommendation
Prognosis
Prognosis depends heavily on underlying aetiology:
| Aetiology | Outlook |
|---|---|
| HIE | ~50% with neurodevelopmental impairment; improved with cooling |
| Hypoglycaemia (treated promptly) | Good if treated within 2 hours |
| Stroke | Depends on territory; epilepsy risk 30-50% |
| Benign familial neonatal seizures | Excellent |
| Ohtahara/EIEE | Poor; often evolves to West syndrome |
| Pyridoxine dependency (treated) | Good if recognised early |
| Congenital malformations | Usually poor; refractory epilepsy |
Neonatal seizures - especially frequent, intractable, or prolonged ones - are independently associated with:
- Further hypoxic-ischaemic brain injury (MR spectroscopy evidence)
- Suppression of neuronal stem cell proliferation
- Later neurodevelopmental impairment (cognitive, motor, epilepsy)
This is why treating even subclinical (electrographic-only) seizures to achieve a lower seizure burden is now recommended.
Summary Algorithm
Neonate with suspected seizure
|
Confirm on EEG (video-EEG or aEEG)
|
Immediate: Check glucose, electrolytes (Ca, Mg, Na)
Treat reversible cause FIRST
|
Blood/CSF cultures → Empiric antibiotics + acyclovir
|
Imaging: cranial US → MRI brain
|
Still seizing?
|
PHENOBARBITAL 20 mg/kg IV (1st line)
|
No response → Phenytoin OR Levetiracetam OR Midazolam
|
Still refractory?
→ Pyridoxine 100 mg IV (with EEG)
→ PLP, folinic acid
→ Genetic testing (gene panel)
|
Seizures resolved - stop ASMs before discharge
(if no ongoing epilepsy)
Sources:
- Bradley and Daroff's Neurology in Clinical Practice - Neonatal Seizures (Chapter 110)
- Adams and Victor's Principles of Neurology, 12th ed. - Neonatal Seizures
- Tintinalli's Emergency Medicine - Neonatal Seizures
- Henry's Clinical Diagnosis and Management by Laboratory Methods
- 2023 ILAE Task Force Guidelines: Pressler RM et al., Epilepsia 2023 - Treatment of seizures in the neonate (evidence-based recommendations)
- Cochrane Review: Abiramalatha T et al., Cochrane Database Syst Rev 2023 (PMID 37873971) - Anti-seizure medications for neonates
This is a shared conversation. Sign in to Orris to start your own chat.