Cll
Reading File
Finding Sources
Searching PubMed
"chronic lymphocytic leukemia" AND treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Excellent - I now have comprehensive information from multiple authoritative sources. Let me compile the full answer:
Chronic Lymphocytic Leukemia (CLL)
Definition
CLL is a monoclonal proliferation of mature, immunophenotypically characteristic B lymphocytes. Diagnosis requires an absolute lymphocyte count of >5 × 10⁹/L of clonal B cells in the peripheral blood with characteristic immunophenotype. When the same cell type is predominantly tissue-based (lymph nodes) without significant blood/marrow involvement, it is called Small Lymphocytic Lymphoma (SLL) - but genetically and molecularly, CLL and SLL are the same entity.
- Harrison's Principles of Internal Medicine 22E, p. 893
- Goldman-Cecil Medicine, p. 1928
Epidemiology
- Most common leukemia in the Western Hemisphere (~20,000+ new US cases/year)
- Median age at diagnosis: 70-72 years
- Male:Female ratio = 2:1 (becomes more equal after age 80)
- More common in Caucasians; rare in Asians/Pacific Islanders even after migration
- 5-year survival improved from 70% (1980) to 92% (2015) and continues to rise
- Strong familial risk: first-degree relatives have 5-8x increased risk; two affected first-degree relatives confer ~27x increased risk
- Associated with Agent Orange/burn pit exposure (service-connected illness for Vietnam veterans)
- NOT linked to radiation exposure (unlike most other leukemias)
Precursor: Monoclonal B-Cell Lymphocytosis (MBL)
MBL is thought to precede every case of CLL. It is defined as CLL-like clonal B cells <5 × 10⁹/L without lymphadenopathy, organomegaly, or cytopenias.
- Low-count MBL (<0.5 × 10⁹/L): negligible risk of progression
- High-count MBL (0.5-5 × 10⁹/L): progresses to CLL at 1-2% per year
- MBL prevalence is ~12% in general population, ~18% in first-degree relatives of CLL patients
Pathology & Immunophenotype
Morphology: Diffuse infiltrate of small mature lymphocytes with clumped chromatin and scant cytoplasm. "Smudge cells" are characteristic on peripheral smear. Proliferation centers (pseudofollicles) containing prolymphocytes are pathognomonic.
Characteristic Immunophenotype (KEY for diagnosis and differential):
| Marker | CLL/SLL |
|---|---|
| CD19, CD20 (dim), CD79a | Positive (B-cell markers) |
| CD5 | Positive (aberrant T-cell marker co-expression) |
| CD23 | Positive |
| CD200 | Positive |
| LEF1 | Positive |
| Cyclin D1, CD10, BCL-6 | Negative |
| Surface Ig | Dim (single light chain) |
CD5+/CD23+ distinguishes CLL from mantle cell lymphoma (CD5+/cyclin D1+/CD23-) and follicular lymphoma (CD10+/BCL-6+).
- Henry's Clinical Diagnosis and Management by Laboratory Methods, p. 3954
Cytogenetics and Molecular Prognostic Markers
Detected by FISH (not routine karyotype - CLL cells are minimally proliferative):
| Abnormality | Frequency | Prognosis |
|---|---|---|
| del(13q14.3) | ~50% | Favorable |
| Trisomy 12 | ~20% | Intermediate |
| del(11q22-23) [ATM] | ~20% | Adverse |
| del(17p13) [TP53] | ~10% | Adverse (high-risk) |
| del(6q21) | ~5% | Adverse |
Other adverse markers:
- Unmutated IGHV (immunoglobulin heavy-chain variable region) - in ~50% of cases; associated with ZAP-70 expression, CD38 expression, and worse prognosis
- TP53 mutation/del(17p): resistance to chemoimmunotherapy
- NOTCH1, SF3B1, BIRC3 mutations: additional adverse features
- β₂-microglobulin elevation, lymphocyte doubling time <12 months: adverse
Staging
Two classical systems:
Rai Staging (US):
| Stage | Findings | Risk |
|---|---|---|
| 0 | Lymphocytosis only | Low |
| I | + Lymphadenopathy | Low |
| II | + Splenomegaly/hepatomegaly | Intermediate |
| III | + Hgb <11 g/dL | High |
| IV | + Platelets <100,000/µL | High |
Binet Staging (Europe):
| Stage | Findings |
|---|---|
| A | <3 lymphoid areas involved |
| B | ≥3 areas involved |
| C | Hgb <10 g/dL OR platelets <100,000/µL |
- Washington Manual of Medical Therapeutics, p. 867
Clinical Presentation
- Most patients are asymptomatic at diagnosis, found incidentally on CBC
- When symptomatic: fatigue, weight loss, night sweats, lymphadenopathy
- Infections (leading cause of morbidity/mortality; 30-50% of deaths)
- Autoimmune complications: Autoimmune hemolytic anemia (AIHA), immune thrombocytopenia (ITP)
- Richter transformation: CLL transforms to diffuse large B-cell lymphoma (DLBCL) in ~5% - presents with rapid nodal enlargement, B symptoms, elevated LDH
Treatment Indications
Watch-and-wait is appropriate for early-stage asymptomatic disease. Treatment is indicated (per IWCLL criteria) when at least one of the following is present:
- Progressive marrow failure (worsening anemia/thrombocytopenia)
- Massive or symptomatic splenomegaly (>6 cm below costal margin)
- Lymphadenopathy >10 cm in longest diameter
- Progressive lymphocytosis (>50% increase in 2 months or doubling time <6 months)
- Symptomatic disease (significant fatigue, night sweats, weight loss, fever)
- AIHA or ITP poorly responsive to steroids
Treatment
Modern therapy is dominated by targeted agents - chemoimmunotherapy is now largely reserved for fit patients without high-risk genetics.
BTK Inhibitors (first-line and relapsed/refractory):
- Ibrutinib - first-generation; once daily; continuous therapy
- Acalabrutinib - more selective, fewer off-target effects (preferred over ibrutinib for cardiac patients)
- Zanubrutinib - most selective BTK inhibitor; shown superior to ibrutinib in relapsed CLL
BCL-2 Inhibitor:
- Venetoclax - used in combination with obinutuzumab (anti-CD20) as fixed-duration therapy; also with ibrutinib or rituximab
For del(17p)/TP53 mutation (high-risk): BTK inhibitors or venetoclax-based regimens preferred (resistant to chemoimmunotherapy)
Chemoimmunotherapy (now less common):
- FCR (fludarabine, cyclophosphamide, rituximab) - still used for young, fit patients with mutated IGHV (potential for very long-term remission)
Recent evidence from a 2025 network meta-analysis (JNCI, PMID 39392788) comparing first-line CLL treatments, and a 2025 network meta-analysis on BTK inhibitors in high-risk relapsed/refractory CLL (Blood Advances, PMID 40203277), continue to refine the optimal sequencing of these agents.
Key Complications Summary
| Complication | Notes |
|---|---|
| Infections | Bacterial (encapsulated organisms), viral (HSV, VZV), fungal |
| AIHA/ITP | Warm antibody AIHA most common; may flare with therapy |
| Hypogammaglobulinemia | Consider IVIG for recurrent infections |
| Richter transformation | Very poor prognosis; treat like aggressive lymphoma |
| Secondary malignancies | Increased risk (skin cancers, other solid tumors) |
Sources:
- Harrison's Principles of Internal Medicine 22E (2025), Ch. 112
- Goldman-Cecil Medicine International Edition, Ch. on CLL
- Washington Manual of Medical Therapeutics, p. 867
- Henry's Clinical Diagnosis and Management by Laboratory Methods, p. 3954
Cml
Finding Sources
Searching PubMed
"chronic myeloid leukemia" AND treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have everything needed for a comprehensive answer.
Chronic Myeloid Leukemia (CML)
Definition
CML is a clonal hematopoietic myeloproliferative stem cell neoplasm driven by the BCR::ABL1 chimeric fusion gene. This results from a reciprocal balanced translocation between the long arms of chromosomes 9 and 22 - t(9;22)(q34.1;q11.2) - producing the famous Philadelphia (Ph) chromosome (shortened chromosome 22). The BCR-ABL fusion protein is a 210 kDa constitutively active tyrosine kinase that drives growth factor-independent proliferation while preserving differentiation.
There is no BCR-ABL-negative CML - what is called "atypical CML" or chronic neutrophilic leukemia are distinct entities with separate genetic features.
- Goldman-Cecil Medicine, p. 1938
- Robbins Pathologic Basis of Disease
Pathogenesis
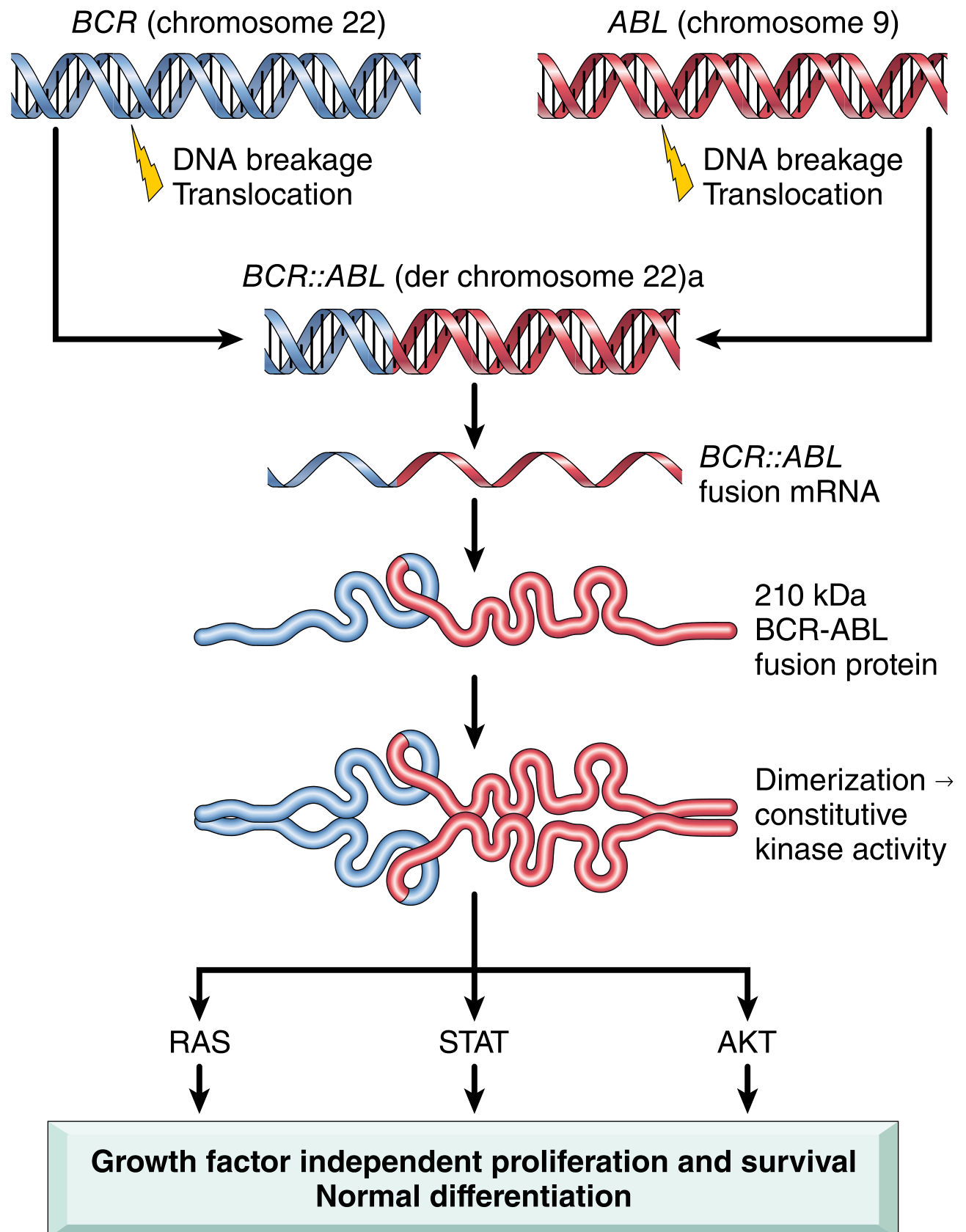
Robbins Pathologic Basis of Disease - Fig. 13.34: BCR (chr 22) and ABL (chr 9) break and fuse → BCR::ABL fusion mRNA → 210 kDa protein → dimerization → constitutive kinase activity → activates RAS, STAT, and AKT pathways → growth factor-independent proliferation with preserved differentiation (key feature - distinguishes from acute leukemia).
Epidemiology
- Accounts for ~14-15% of all leukemias
- Annual incidence: ~2 per 100,000; ~8,000-9,000 new US cases/year
- Median age at diagnosis: 55-65 years (younger than CLL)
- Slight male predominance (M:F = 1.6:1)
- Uncommon in children (<3% of CML cases under age 20)
- Risk factors: ionizing radiation (unlike CLL); no consistent links to chemical exposures
Prognosis transformation with TKIs:
- Pre-TKI era: median survival 3-7 years; 10-year survival <30%
- TKI era (imatinib): 10-year survival >85%, approaching age-matched normal population
Clinical Phases
CML follows a triphasic (or biphasic) course:
1. Chronic Phase (~75% at diagnosis)
- Insidious onset; most patients found incidentally on routine CBC
- Symptoms when present: fatigue, weight loss, anorexia, splenomegaly (left upper quadrant dragging pain, early satiety, splenic infarction)
- Peripheral blood: marked leukocytosis (often >100,000 cells/µL) with the full granulocytic spectrum - neutrophils, band forms, metamyelocytes, myelocytes, eosinophils, basophilia (characteristic)
- Blasts typically <10% in blood
- Platelets often elevated (sometimes markedly)
- Bone marrow: markedly hypercellular, granulocytic hyperplasia, "sea-blue histiocytes" (scattered macrophages with green-blue cytoplasm), increased reticulin
2. Accelerated Phase
- Worsening anemia and thrombocytopenia, rising basophils
- Additional clonal cytogenetic abnormalities (trisomy 8, isochromosome 17q, duplication of Ph chromosome)
- ELN criteria: blasts 15-<30% in blood or marrow
- WHO criteria: blasts 10-20%
3. Blast Phase (Blast Crisis)
- Resembles acute leukemia
- ELN criteria: blasts ≥30%; WHO criteria: blasts ≥20%
- 70% myeloid blast crisis, ~30% lymphoid blast crisis (pre-B cell) - evidence that CML originates from a pluripotent stem cell
- Prognosis poor; modest benefit from TKIs in blast phase
About 50% of untreated chronic-phase patients progress through an accelerated phase before blast crisis; the other 50% go directly to blast crisis.
Diagnosis
| Test | Finding |
|---|---|
| CBC + differential | Leukocytosis with left shift, basophilia, eosinophilia |
| Bone marrow biopsy | Hypercellular, granulocytic hyperplasia, ↑reticulin |
| Cytogenetics (karyotype) | Philadelphia chromosome t(9;22) in >90% |
| FISH | Detects BCR-ABL in cytogenetically cryptic cases |
| RT-PCR (qPCR) | Detects and quantifies BCR-ABL1 transcripts (used for monitoring) |
| LAP (leukocyte alkaline phosphatase) | LOW in CML (distinguishes from leukemoid reaction where LAP is high) |
Staging / Response Criteria
Monitored by quantitative PCR (qPCR) of BCR-ABL1 transcripts on the International Scale (IS):
| Response | Definition |
|---|---|
| Complete Hematologic Response (CHR) | Normalization of CBC + no splenomegaly |
| Complete Cytogenetic Response (CCyR) | No Ph+ metaphases on marrow cytogenetics |
| Major Molecular Response (MMR) | BCR-ABL1 ≤0.1% (IS) = 3-log reduction |
| Deep Molecular Response (MR4/MR4.5) | BCR-ABL1 ≤0.01% or ≤0.0032% (IS) |
Optimal response milestones (ELN):
- By 3 months: BCR-ABL1 ≤10% (IS)
- By 6 months: BCR-ABL1 ≤1% (IS) / CCyR
- By 12 months: BCR-ABL1 ≤0.1% (IS) = MMR
Failure to achieve milestones → consider TKI switch + test for kinase domain resistance mutations.
TKI Therapy - The Cornerstone of Treatment
Six FDA-approved oral BCR-ABL1 TKIs:
| Drug | Generation | Dose | Notable Toxicities |
|---|---|---|---|
| Imatinib (Gleevec) | 1st | 400 mg daily | Edema, nausea, muscle cramps, rash |
| Dasatinib (Sprycel) | 2nd | 100 mg daily (1L) | Pleural/pericardial effusions, pulmonary hypertension, myelosuppression |
| Nilotinib (Tasigna) | 2nd | 300 mg BID | Cardiovascular/arterial occlusive events, QTc prolongation, pancreatitis |
| Bosutinib (Bosulif) | 2nd | 400 mg daily | Diarrhea, hepatotoxicity |
| Ponatinib (Iclusig) | 3rd | Variable | Hypertension, arterial thrombosis (inhibits VEGFR), pancreatitis |
| Asciminib (Scemblix) | 3rd (STAMP inhibitor) | 40 mg BID or 200 mg BID (T315I) | Myelosuppression, hypertension |
Key points:
- Nilotinib is 30x more potent than imatinib (structurally similar)
- Dasatinib is ~300x more potent; also inhibits SRC kinases
- Bosutinib is 30-50x more potent; also inhibits SRC kinases
- Ponatinib and asciminib are active against the T315I "gatekeeper" mutation (resistant to all other TKIs)
- Asciminib is a STAMP inhibitor - uniquely targets the ABL myristoyl pocket (allosteric inhibition) rather than the ATP binding site - a different mechanism from all other TKIs
- Asciminib in newly diagnosed CML (ASCEMBL trial data, NEJM 2024) shows promising results as 1st-line therapy
Choice of first-line agent depends on patient comorbidities, toxicity profile, and goal (some patients aim for treatment-free remission, requiring deeper responses earlier).
TKI Resistance and Mutations
When patients fail TKI therapy, test for BCR-ABL1 kinase domain mutations:
- Most common resistance mutations: E255K, Y253H (nilotinib/dasatinib sensitive mutations)
- T315I ("gatekeeper mutation"): confers resistance to all 1st and 2nd generation TKIs → use ponatinib or asciminib
Other options for TKI resistance: omacetaxine mepesuccinate (protein synthesis inhibitor; activity independent of BCR-ABL mutation status).
Treatment-Free Remission (TFR)
A key modern goal - patients who achieve deep and sustained molecular responses (MR4.5) may attempt TKI discontinuation:
- ~40-60% of eligible patients maintain TFR
- Most relapses occur within the first 6 months and respond to TKI rechallenge
- Requires close molecular monitoring after stopping
The EURO-SKI trial final analysis (JCO 2024) has helped define optimal TFR criteria.
Management of Advanced Phase / Blast Crisis
- Accelerated phase: TKIs (often higher dose or 2nd/3rd generation) ± consideration of allogeneic HSCT
- Blast crisis: TKI + acute leukemia-type chemotherapy (regimen depends on myeloid vs. lymphoid crisis) → bridge to allogeneic HSCT if response achieved
- HSCT is now a later-line option after TKI failure; no longer first-line for chronic phase
CML vs. Leukemoid Reaction (key differential)
| Feature | CML | Leukemoid Reaction |
|---|---|---|
| LAP score | Low | High |
| Basophilia | Present | Absent |
| Philadelphia chromosome | Present | Absent |
| BCR-ABL1 | Positive | Negative |
| Splenomegaly | Often marked | Usually absent |
Sources:
- Harrison's Principles of Internal Medicine 22E (2025), Ch. 110
- Goldman-Cecil Medicine International Edition, Ch. on CML
- Robbins Pathologic Basis of Disease, Ch. 13
- Washington Manual of Medical Therapeutics, p. 866
Recent evidence: A 2024-2025 systematic review comparing ponatinib vs. asciminib in post-2nd-generation TKI therapy continues to refine the optimal sequencing in resistant CML. A 2025 meta-analysis on TKI toxicity (Support Care Cancer, PMID 40317312) highlights the significant symptom burden that affects quality of life and adherence.
Cml
Here is the full CML summary from the previous response - no need to re-research:
Chronic Myeloid Leukemia (CML)
Definition
CML is a clonal hematopoietic myeloproliferative stem cell neoplasm driven by the BCR::ABL1 chimeric fusion gene, resulting from the reciprocal translocation t(9;22)(q34.1;q11.2) - the Philadelphia (Ph) chromosome (shortened chromosome 22). The BCR-ABL fusion protein is a 210 kDa constitutively active tyrosine kinase that drives growth factor-independent proliferation while preserving differentiation.
There is no BCR-ABL-negative CML - "atypical CML" and chronic neutrophilic leukemia are distinct entities.
Pathogenesis
BCR (chr 22) + ABL (chr 9) → BCR::ABL fusion mRNA → 210 kDa protein → constitutive kinase → activates RAS, STAT, AKT → growth factor-independent proliferation with preserved differentiation (unlike acute leukemia).
Epidemiology
- ~14-15% of all leukemias; ~8,000-9,000 new US cases/year
- Median age: 55-65 years; male predominance (M:F = 1.6:1)
- Risk factor: ionizing radiation (unlike CLL)
- Pre-TKI: median survival 3-7 years | TKI era: 10-year survival >85%
Clinical Phases
1. Chronic Phase (~75% at diagnosis)
- Usually asymptomatic, found on routine CBC
- Symptoms: fatigue, weight loss, splenomegaly (LUQ pain, early satiety)
- Blood: leukocytosis often >100,000/µL, full granulocytic spectrum, basophilia (characteristic), eosinophilia, thrombocytosis
- Blasts <10%; bone marrow hypercellular with "sea-blue histiocytes"
2. Accelerated Phase
- Worsening anemia/thrombocytopenia, rising basophils
- Additional cytogenetic abnormalities (trisomy 8, iso17q, Ph duplication)
- ELN: blasts 15-<30% | WHO: blasts 10-20%
3. Blast Phase (Blast Crisis)
- Resembles acute leukemia
- ELN: blasts ≥30% | WHO: blasts ≥20%
- 70% myeloid, ~30% lymphoid (pre-B cell) - confirms pluripotent stem cell origin
- 50% progress through accelerated phase; 50% go directly to blast crisis
Diagnosis
| Test | Finding |
|---|---|
| CBC | Leukocytosis, basophilia, left shift |
| Bone marrow biopsy | Hypercellular, granulocytic hyperplasia |
| Karyotype | Ph chromosome t(9;22) in >90% |
| FISH | Detects cryptic BCR-ABL rearrangements |
| qPCR | Quantifies BCR-ABL1 transcripts (used for monitoring) |
| LAP score | LOW (distinguishes from leukemoid reaction) |
Treatment Response Milestones
| Timepoint | Target (BCR-ABL1 IS) |
|---|---|
| 3 months | ≤10% |
| 6 months | ≤1% (CCyR) |
| 12 months | ≤0.1% = MMR |
- CHR: normalized CBC + no splenomegaly
- CCyR: no Ph+ metaphases on marrow cytogenetics
- MMR (MR3): BCR-ABL1 ≤0.1% IS
- Deep MR (MR4/MR4.5): ≤0.01% / ≤0.0032% IS - required for TFR attempt
FDA-Approved TKIs
| Drug | Generation | Dose | Notable Toxicities |
|---|---|---|---|
| Imatinib (Gleevec) | 1st | 400 mg daily | Edema, nausea, cramps |
| Dasatinib (Sprycel) | 2nd | 100 mg daily | Pleural/pericardial effusion, pulmonary HTN |
| Nilotinib (Tasigna) | 2nd | 300 mg BID | Arterial occlusion, QTc prolongation |
| Bosutinib (Bosulif) | 2nd | 400 mg daily | Diarrhea, hepatotoxicity |
| Ponatinib (Iclusig) | 3rd | Variable | Arterial thrombosis, hypertension (inhibits VEGFR) |
| Asciminib (Scemblix) | 3rd (STAMP) | 40 mg BID | Myelosuppression, hypertension |
Key points:
- Nilotinib = 30x more potent than imatinib
- Dasatinib = ~300x more potent; also inhibits SRC kinases
- Ponatinib + Asciminib = active against T315I "gatekeeper" mutation
- Asciminib uniquely targets the ABL myristoyl pocket (allosteric) - different from all other TKIs
- First-line choice guided by comorbidities, toxicity profile, and TFR goal
TKI Resistance
- Test for BCR-ABL1 kinase domain mutations when milestones not met
- T315I mutation → resistant to all 1st/2nd gen TKIs → use ponatinib or asciminib
- Omacetaxine (protein synthesis inhibitor): option when TKI resistance is BCR-ABL mutation-independent
Treatment-Free Remission (TFR)
- Patients achieving sustained MR4.5 may attempt TKI discontinuation
- ~40-60% maintain TFR; most relapses within 6 months and respond to TKI rechallenge
- Requires close molecular monitoring
Advanced Phase Management
- Accelerated phase: 2nd/3rd gen TKI ± allogeneic HSCT consideration
- Blast crisis: TKI + acute leukemia chemotherapy (myeloid vs. lymphoid regimen) → bridge to allogeneic HSCT
- HSCT is now later-line (after TKI failure), not first-line for chronic phase
CML vs. Leukemoid Reaction
| Feature | CML | Leukemoid Reaction |
|---|---|---|
| LAP score | Low | High |
| Basophilia | Present | Absent |
| Philadelphia chromosome | Present | Absent |
| Splenomegaly | Often marked | Usually absent |
Sources: Harrison's 22E, Goldman-Cecil Medicine, Robbins Pathologic Basis of Disease, Washington Manual of Medical Therapeutics
Make 30 vignettes clinical scenario based on myeloprolifertive syndromes at final yr level covering every aspect of all topics with explanating the right and wrong answers
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Quick Reference: Topics Covered Across the 30 Vignettes
| # | Topic | Key Concept Tested |
|---|---|---|
| 1 | CML Diagnosis | Classic CBC + low LAP + splenomegaly |
| 2 | CML Molecular Test | BCR-ABL1 qPCR as diagnostic/monitoring tool |
| 3 | CML Response Milestones | ELN milestones at 3/6/12 months |
| 4 | T315I Resistance | Ponatinib/asciminib as only active agents |
| 5 | CML Blast Crisis | Myeloid vs lymphoid; HSCT role |
| 6 | Asciminib Mechanism | STAMP inhibitor/myristoyl pocket |
| 7 | PV Diagnosis | JAK2 V617F + low EPO + aquagenic pruritus |
| 8 | PV vs Secondary Polycythemia | EPO level as discriminator |
| 9 | PV Complication | Budd-Chiari syndrome |
| 10 | PV Treatment | Phlebotomy + HU + aspirin (high-risk) |
| 11 | PV Spent Phase | Post-PV myelofibrosis |
| 12 | ET Diagnosis | JAK2-negative → test CALR/MPL |
| 13 | ET Bleeding | Acquired von Willebrand disease with extreme thrombocytosis |
| 14 | ET Risk Stratification | Low-risk: observation + aspirin only |
| 15 | Erythromelalgia | Aspirin-responsive microvascular thrombosis |
| 16 | PMF Morphology | Teardrop cells + leukoerythroblastic picture + fibrosis |
| 17 | PMF Molecular Markers | JAK2 negative → CALR/MPL testing |
| 18 | PMF Prognostication | IPSS scoring system |
| 19 | PMF Treatment | Ruxolitinib (JAK1/2 inhibitor) |
| 20 | PMF Osteosclerosis | Megakaryocyte cytokine-driven bone changes |
| 21 | MPN Differentiation | PV vs CML comparison |
| 22 | Ph+ vs Ph- MPNs | CML only is Ph-positive |
| 23 | Hydroxyurea Toxicity | Leg ulcers/macrocytosis → ruxolitinib/IFN-α |
| 24 | PMF vs CML | BCR-ABL1 as key differentiator |
| 25 | Reactive vs Clonal Thrombocytosis | Splenectomy + iron deficiency |
| 26 | MPN → AML Transformation | Post-PV AML; poor prognosis |
| 27 | CML in Pregnancy | TKIs teratogenic; use IFN-α |
| 28 | Dasatinib Side Effects | Pleural effusion; hold + manage |
| 29 | Treatment-Free Remission | MR4.5 ≥2 years; ~50% maintain TFR |
| 30 | Integrated PV Scenario | Iron deficiency masking PV + HU indication + erythromelalgia |
Sources: Harrison's Principles of Internal Medicine 22E, Robbins Pathologic Basis of Disease, Goldman-Cecil Medicine, Washington Manual of Medical Therapeutics, Henry's Clinical Diagnosis and Management by Laboratory Methods
Make 30 clinical based vignettes on polycythemia vera essential thrombocytopenia covering every aspect of it with explanation of right and wrong ones with gap between question and answer
Reading File
Reading File
Reading File
Reading File
Reading File
30 Clinical Vignettes: Polycythemia Vera & Essential Thrombocythemia
Final Year Level | Complete Answer Explanations
PART 1: POLYCYTHEMIA VERA (15 Vignettes)
Vignette 1 - Classic Presentation
A 64-year-old man presents with a 6-month history of headaches, facial redness, and itching that occurs every time he has a hot shower. He reports no cough or fever. On examination: BP 168/96 mmHg, facial plethora, conjunctival injection, splenomegaly. CBC: Hgb 19.9 g/dL, Hct 62%, WBC 13,400/µL, platelets 560,000/µL. Serum EPO is low.
What is the most likely diagnosis?
A) Secondary polycythemia due to sleep apnea
B) Polycythemia vera
C) Essential thrombocythemia
D) Chronic myeloid leukemia
E) Relative polycythemia (Gaisbock syndrome)
✅ ANSWER: B - Polycythemia Vera
Why B is correct:
The triad of elevated Hgb (>16.5 g/dL in men), panmyelosis (raised WBC + platelets), and low serum EPO is the hallmark of PV. The JAK2 V617F mutation makes erythroid progenitors EPO-independent, so the feedback loop suppresses EPO secretion. "Aquagenic pruritus" (itching after warm water contact) is caused by histamine release from basophils in the neoplastic clone - it is so characteristic of PV that it is essentially pathognomonic. Facial plethora, hypertension, and splenomegaly from vascular congestion complete the picture.
Why others are wrong:
- ❌ A (Sleep apnea/secondary polycythemia): Secondary causes have elevated EPO (compensatory response to hypoxia) and isolated RBC elevation - WBC and platelets are normal. Aquagenic pruritus does not occur.
- ❌ C (ET): ET has markedly elevated platelets as the dominant feature; Hgb/Hct are normal.
- ❌ D (CML): CML presents with extreme leukocytosis (WBC often >100,000) with the full granulocytic spectrum, basophilia, and BCR-ABL1 positivity. The EPO is not the key marker.
- ❌ E (Relative polycythemia): Relative/spurious polycythemia occurs due to plasma volume contraction (dehydration, diuretics). WBC and platelets are normal; EPO is normal; no aquagenic pruritus or splenomegaly.
Vignette 2 - PV Diagnostic Criteria
A 58-year-old woman has Hgb 17.2 g/dL (diagnostic threshold in women is >16.0 g/dL), WBC 12,000/µL, platelets 490,000/µL. Serum EPO is below normal range. JAK2 V617F returns positive.
According to WHO/ICC criteria, has she met the diagnostic threshold for PV?
A) No - bone marrow biopsy showing panmyelosis is always mandatory first
B) Yes - two major criteria are met (elevated Hgb + JAK2 V617F), which is sufficient for diagnosis
C) No - JAK2 alone is not specific enough for PV
D) Yes, but only if BCR-ABL1 is also negative
E) No - EPO must be normal to diagnose PV
✅ ANSWER: B - Yes, two major criteria are sufficient
Why B is correct:
The WHO/ICC diagnostic criteria for PV:
- Major criteria: (1) Hgb >16.5 g/dL (M) or >16.0 g/dL (F), or Hct >49% (M) or >48% (F); (2) Bone marrow hypercellularity with panmyelosis; (3) JAK2 V617F or JAK2 exon 12 mutation
- Minor criterion: Subnormal serum EPO
Diagnosis = all 3 major criteria, OR first 2 major criteria + minor criterion (low EPO). Here: Hgb >16.0 in a woman (major #1) + JAK2 V617F (major #3) + low EPO (minor) = diagnosis established. Bone marrow is not always required if molecular + laboratory criteria are met.
Why others are wrong:
- ❌ A: Bone marrow biopsy is required when Hgb/Hct does not reach the threshold but JAK2 is positive, or when polycythemia vera is suspected without a clear molecular marker. It is not always mandatory first.
- ❌ C: JAK2 V617F is present in >97% of PV; it is highly specific for clonal erythrocytosis in the right context.
- ❌ D: BCR-ABL1 exclusion is part of the PV workup, but it is not a criterion that must be stated as positive/negative to make the PV diagnosis. Excluding CML is implicit.
- ❌ E: Low EPO supports the diagnosis - it is a minor criterion. A normal EPO would argue against PV (or at least require further workup), but low EPO is not required if all 3 major criteria are met.
Vignette 3 - PV Pathogenesis
A medical student asks why patients with polycythemia vera have low serum EPO despite having severe anemia-driven symptoms in other conditions with high EPO levels.
What is the mechanism responsible for low EPO in PV?
A) PV damages the kidneys which produce EPO
B) JAK2 V617F mutation causes constitutive activation of the JAK-STAT pathway, making erythroid progenitors EPO-independent; negative feedback suppresses EPO production
C) High hemoglobin causes EPO receptor downregulation
D) Bone marrow fibrosis prevents EPO from reaching progenitor cells
E) Anti-EPO autoantibodies are produced in PV
✅ ANSWER: B - JAK2 V617F → constitutive JAK-STAT activation → EPO-independent proliferation → suppressed EPO
Why B is correct:
JAK2 V617F (valine-to-phenylalanine substitution at residue 617) constitutively activates the JAK2 tyrosine kinase, which lies downstream of the erythropoietin receptor (and other growth factor receptors including thrombopoietin and G-CSF receptors). Because the JAK-STAT pathway is permanently "switched on" regardless of EPO binding, erythroid progenitors proliferate autonomously. The resulting high RBC mass is detected by the kidneys via oxygen sensors, which then suppress EPO secretion via normal negative feedback. This low EPO is the physiological fingerprint of clonal (primary) versus reactive (secondary) erythrocytosis.
Why others are wrong:
- ❌ A: PV does not directly damage the kidneys. Renal involvement may occur from hyperviscosity complications, but this is not the mechanism of low EPO.
- ❌ C: EPO receptor downregulation from high Hgb is not the established mechanism in PV.
- ❌ D: Bone marrow fibrosis is a late complication of PV (spent phase), not the mechanism of low EPO at diagnosis.
- ❌ E: Anti-EPO antibodies cause pure red cell aplasia (low Hgb), the opposite of PV.
Vignette 4 - PV vs Relative Polycythemia
A 45-year-old man is found to have Hct 54% on routine labs. He is obese, smokes heavily, and drinks alcohol daily. He denies pruritus. BP is normal. No splenomegaly. WBC and platelets are normal. EPO level is normal. JAK2 V617F is negative.
What is the most likely diagnosis?
A) Polycythemia vera
B) Relative/spurious polycythemia (Gaisbock syndrome)
C) Secondary polycythemia from COPD
D) Essential thrombocythemia with masked polycythemia
E) Chuvash polycythemia
✅ ANSWER: B - Relative/Spurious Polycythemia (Gaisbock Syndrome)
Why B is correct:
Relative (spurious) polycythemia occurs when the plasma volume is reduced (hemoconcentration) while the total RBC mass is normal. Classic risk factors include obesity, heavy smoking, hypertension, chronic alcohol use, and stress. Key features: isolated elevated Hct with normal WBC, normal platelets, normal EPO, and negative JAK2. No splenomegaly. No aquagenic pruritus. Treatment targets the underlying risk factors (smoking cessation, hydration, weight loss, blood pressure control).
Why others are wrong:
- ❌ A (PV): JAK2 V617F is negative and EPO is normal. PV requires JAK2 positivity or low EPO + bone marrow panmyelosis.
- ❌ C (Secondary COPD): Secondary polycythemia from COPD would have elevated EPO and may show signs of chronic lung disease (cor pulmonale, reduced spirometry). It is possible in a smoker, but normal EPO + normal WBC/platelets in a plethoric obese/alcohol-drinking patient with no pulmonary symptoms points to relative polycythemia.
- ❌ D: ET does not cause elevated Hct as a primary feature.
- ❌ E (Chuvash polycythemia): This is a rare autosomal recessive condition due to VHL mutations causing high-affinity EPO sensing. It is congenital, typically presents in childhood/young adulthood, and EPO is elevated - not the picture here.
Vignette 5 - PV and Thrombosis
A 67-year-old woman with known PV presents to the emergency department with sudden onset right arm weakness and expressive aphasia lasting 45 minutes, followed by complete resolution. CT head is normal.
What most likely caused this event, and what PV-related factor is primarily responsible?
A) Hemorrhagic stroke from thrombocytopenia
B) Transient ischemic attack (TIA) from increased blood viscosity and hyperviscosity-related thromboembolism in PV
C) Paradoxical embolism through a patent foramen ovale
D) Atrial fibrillation-related cardioembolic TIA, unrelated to PV
E) Hyperviscosity syndrome from IgM paraprotein
✅ ANSWER: B - TIA from PV-related hyperviscosity and thrombosis
Why B is correct:
Neurological events (TIA, stroke) are among the most common life-threatening complications of PV, occurring in ~30% of patients. The mechanism is multifactorial:
- Increased blood viscosity from elevated RBC mass → sluggish flow and vascular sludging
- Thrombocytosis with dysfunctional platelets → paradoxical tendency to both thrombose and bleed
- Basophilia and histamine contribute to vascular reactivity
This TIA is a high-risk feature of PV (age >60 + prior thrombotic event), mandating cytoreductive therapy (hydroxyurea) in addition to phlebotomy and aspirin.
Why others are wrong:
- ❌ A: PV causes thrombocytosis (elevated platelets), not thrombocytopenia. Hemorrhagic events do occur but TIAs are thrombotic.
- ❌ C: PFO-related paradoxical emboli require a venous source. While possible, this ignores the direct PV pathophysiology.
- ❌ D: While AF is common in the elderly, this patient's PV with elevated viscosity is the most likely culprit. PV management must be addressed regardless.
- ❌ E: Hyperviscosity from IgM paraprotein occurs in Waldenström macroglobulinemia (plasma cell/lymphoplasmacytic neoplasm), causing hyperviscosity syndrome with very different lab findings (elevated IgM, normal RBC mass).
Vignette 6 - PV Risk Stratification
Two patients with confirmed PV present for management counseling:
- Patient A: 55 years old, no prior thrombosis, no cardiovascular risk factors
- Patient B: 72 years old, prior DVT 2 years ago, hypertension
How should their treatments differ?
A) Both patients require hydroxyurea and phlebotomy equally
B) Patient A: phlebotomy + aspirin (low-risk); Patient B: phlebotomy + hydroxyurea + aspirin (high-risk)
C) Patient B should go directly to allogeneic HSCT
D) Patient A needs no treatment; Patient B needs phlebotomy alone
E) Both patients should receive ruxolitinib as first-line
✅ ANSWER: B - Risk-stratified treatment
Why B is correct:
PV risk stratification is based on two factors:
- High-risk: Age ≥60 OR history of thrombosis
- Low-risk: Age <60 AND no prior thrombosis
Low-risk PV (Patient A): Phlebotomy (target Hct <45%) + low-dose aspirin (81 mg). No cytoreductive therapy unless counts worsen or symptoms develop.
High-risk PV (Patient B): Meets both criteria (age 72 + prior DVT) → phlebotomy + hydroxyurea (first-line cytoreduction) + aspirin. Cytoreduction reduces thrombotic events significantly.
Why others are wrong:
- ❌ A: Over-treating low-risk PV with hydroxyurea exposes the patient to unnecessary toxicity (myelosuppression, leg ulcers, macrocytosis) without proven additional benefit.
- ❌ C: Allogeneic HSCT is not standard for PV; it is reserved for post-PV myelofibrosis or AML transformation in selected patients.
- ❌ D: No treatment for low-risk is too passive - phlebotomy + aspirin is still required for all PV patients to prevent vascular complications.
- ❌ E: Ruxolitinib is FDA-approved for PV refractory to or intolerant of hydroxyurea - it is second-line, not first-line.
Vignette 7 - PV Treatment Target
A 61-year-old man with PV is being managed with phlebotomy. His physician sets a hematocrit target.
What is the evidence-based hematocrit target for phlebotomy in PV?
A) <50% in all patients
B) <45% in both men and women
C) <55% in men and <50% in women
D) <40% to achieve full normalization
E) Target depends only on symptoms, not a specific Hct value
✅ ANSWER: B - Target Hct <45% in both men and women
Why B is correct:
The CYTO-PV trial (landmark RCT) demonstrated that maintaining Hct <45% versus 45-50% in PV significantly reduced the composite endpoint of cardiovascular death and major thrombotic events (2.7% vs 9.8%, p=0.02). This applies to both men and women. The physiological rationale: blood viscosity rises steeply above Hct 45%, dramatically increasing thrombotic risk. Phlebotomy (therapeutic venesection) is performed every 1-3 days initially until Hct <45% is reached, then periodically to maintain this target.
Why others are wrong:
- ❌ A: A target of <50% was used historically but is now known to be insufficient - the CYTO-PV trial clearly showed <45% is superior.
- ❌ C: There is no sex-based differential in the Hct target for treatment. The diagnostic thresholds differ by sex (>16.5 g/dL M, >16.0 g/dL F) but the treatment target Hct <45% is the same.
- ❌ D: A target of <40% is overly aggressive and risks iron deficiency anemia, fatigue, and reduced quality of life without additional benefit.
- ❌ E: While symptoms guide treatment frequency, the Hct <45% target is evidence-based and should be the objective regardless of symptom status.
Vignette 8 - PV Hydroxyurea Indication and Toxicity
A 69-year-old woman with PV managed on hydroxyurea for 3 years develops painful oral ulcers, non-healing leg ulcers over both malleoli, and macrocytic anemia (MCV 108 fL). Her Hct has dropped to 36%.
What is happening and what is the most appropriate next step?
A) She has developed PV transformation to AML - urgent bone marrow biopsy
B) She has developed hydroxyurea toxicity; switch to ruxolitinib or interferon-alpha
C) The leg ulcers represent venous insufficiency - wound care and compression stockings
D) Macrocytosis indicates B12 deficiency - supplement B12
E) Stop all therapy and observe; the disease has spontaneously resolved
✅ ANSWER: B - Hydroxyurea toxicity; switch to ruxolitinib or interferon-alpha
Why B is correct:
Hydroxyurea toxicity is well-recognized and includes:
- Mucocutaneous ulcers (oral and particularly leg ulcers over malleoli) - a characteristic and treatment-limiting side effect
- Macrocytosis (ribonucleotide reductase inhibition impairs DNA synthesis, mimicking megaloblastic changes)
- Skin hyperpigmentation, nail changes, squamous cell carcinoma risk with prolonged use
- Myelosuppression (anemia, neutropenia)
When toxicity occurs, the patient is classified as hydroxyurea-intolerant. Options:
- Ruxolitinib - FDA-approved for HU-refractory/intolerant PV; reduces Hct, controls symptoms, improves quality of life
- Interferon-alpha (pegylated) - preferred in younger patients, pregnancy; can achieve molecular remission of JAK2 allele burden
Why others are wrong:
- ❌ A (AML transformation): AML presents with cytopenias AND high blast count (≥20%). No mention of blasts here, and the clinical picture is clearly drug toxicity.
- ❌ C: While leg ulcers can occur with venous insufficiency, the combination with oral ulcers and macrocytosis in a patient on long-term hydroxyurea strongly points to drug toxicity, not venous insufficiency alone.
- ❌ D: B12 deficiency causes macrocytosis but NOT leg ulcers in this pattern. The causal relationship to hydroxyurea is clear.
- ❌ E: Stopping all therapy in PV without replacement risks rebound polycythemia and thrombosis.
Vignette 9 - PV and Gout
A 66-year-old man with PV presents with a painful swollen right big toe. Serum uric acid is 11.2 mg/dL. X-ray shows soft tissue tophi. He is currently on phlebotomy and low-dose aspirin only.
Why does PV cause gout, and how should it be managed?
A) PV patients eat a high-purine diet; dietary restriction alone is sufficient
B) High cell turnover from panmyeloid proliferation leads to increased uric acid production; treat with allopurinol
C) Aspirin blocks uric acid excretion causing gout; stop aspirin
D) Gout in PV is coincidental; treat as primary gout with colchicine only
E) Phlebotomy itself releases purines causing gout; reduce phlebotomy frequency
✅ ANSWER: B - High cell turnover → hyperuricemia; treat with allopurinol
Why B is correct:
PV involves panmyeloid proliferation with a high rate of cell turnover. When rapidly dividing cells die, nucleic acids (DNA and RNA) are broken down, releasing purines that are metabolized to uric acid. Symptomatic gout occurs in 5-10% of PV patients. Allopurinol (xanthine oxidase inhibitor) reduces uric acid production and is the treatment of choice for PV-associated hyperuricemia. Adequate hydration and cytoreductive therapy (reducing cell turnover) also help.
Why others are wrong:
- ❌ A: While diet plays a role, the primary mechanism is endogenous uric acid overproduction from cell turnover - dietary restriction alone is insufficient.
- ❌ C: Low-dose aspirin (81 mg) does NOT meaningfully inhibit uric acid excretion (that effect is at higher analgesic/anti-inflammatory doses ≥2-3 g/day). Stopping aspirin would increase thrombotic risk dramatically in PV.
- ❌ D: Gout in PV has a specific mechanism (high cell turnover). Colchicine treats the acute attack, but allopurinol is needed for prevention.
- ❌ E: Phlebotomy does release some cell contents, but it is not the primary driver of gout. Reducing phlebotomy to "manage gout" would be dangerous, allowing Hct to rise and increasing thrombotic risk.
Vignette 10 - PV and Peptic Ulcer Disease
A 60-year-old woman with PV reports a 3-month history of epigastric pain and dyspepsia. Upper GI endoscopy reveals a duodenal ulcer. She denies NSAID use. H. pylori testing is negative.
What is the mechanism linking PV to peptic ulcers?
A) PV causes hypersecretion of gastric acid through direct parietal cell stimulation
B) Elevated RBC mass reduces mucous layer protection
C) Histamine released from basophils in the neoplastic clone stimulates gastric acid secretion
D) Iron deficiency from phlebotomy damages the gastric mucosa
E) Low EPO levels increase susceptibility to H. pylori
✅ ANSWER: C - Histamine from basophils stimulates gastric acid secretion
Why C is correct:
One of the pathognomonic features of PV is basophilia - the neoplastic clone includes basophils which release histamine. Histamine acts on gastric parietal cell H₂ receptors, stimulating acid secretion. This is the same mechanism by which basophilic/mast cell diseases (like systemic mastocytosis) cause peptic ulcers. The histamine also contributes to aquagenic pruritus. In PV, peptic ulcer disease and pruritus are both histamine-mediated phenomena from the same basophilic proliferation. H₂ antagonists or proton pump inhibitors are appropriate adjunctive treatment.
Why others are wrong:
- ❌ A: PV does not directly stimulate parietal cells independent of histamine.
- ❌ B: No direct evidence that elevated RBC mass damages the gastric mucosa.
- ❌ D: Iron deficiency from phlebotomy affects RBC size (microcytosis), not gastric mucosa directly.
- ❌ E: EPO has no known role in H. pylori susceptibility or gastric mucosal protection.
Vignette 11 - PV in Pregnancy
A 31-year-old woman with confirmed PV (JAK2 V617F+, Hgb 18.1 g/dL) presents 8 weeks pregnant. She is currently on phlebotomy alone.
Which of the following is the safest management plan during pregnancy?
A) Start hydroxyurea to control counts - it is safe in pregnancy
B) Continue phlebotomy and add interferon-alpha if cytoreduction is needed; avoid hydroxyurea and aspirin entirely
C) Continue phlebotomy + low-dose aspirin; add interferon-alpha if cytoreduction needed; avoid hydroxyurea
D) Terminate the pregnancy as PV is a contraindication to pregnancy
E) Perform therapeutic apheresis (erythrocytapheresis) weekly throughout pregnancy
✅ ANSWER: C - Phlebotomy + low-dose aspirin + interferon-alpha if needed; avoid hydroxyurea
Why C is correct:
Management of PV in pregnancy:
- Phlebotomy is safe throughout pregnancy (target Hct <45%)
- Low-dose aspirin (81 mg) is recommended - reduces placental thrombosis risk (category B; used safely in pregnancy for preeclampsia prophylaxis)
- Hydroxyurea is teratogenic (causes fetal malformations - limb defects, neural tube defects) and is absolutely contraindicated in pregnancy
- Interferon-alpha does not cross the placenta (large molecule), is safe in pregnancy, and is the preferred cytoreductive agent when needed
- PV in pregnancy carries high-risk for both maternal (thrombosis, hemorrhage) and fetal complications (miscarriage, growth restriction)
Why others are wrong:
- ❌ A: Hydroxyurea is teratogenic (FDA Category D) - CONTRAINDICATED in pregnancy. This is a critical safety point.
- ❌ B: Low-dose aspirin is actually recommended in PV pregnancy to reduce thrombotic complications; the statement to avoid it entirely is incorrect.
- ❌ D: Pregnancy in PV is high-risk but not contraindicated. With appropriate management (multidisciplinary care), successful outcomes are achievable.
- ❌ E: Erythrocytapheresis may be used in selected cases but is not routine weekly management for PV in pregnancy.
Vignette 12 - Post-PV Myelofibrosis
A 74-year-old man with a 14-year history of PV now presents with increasing fatigue, massive splenomegaly (22 cm), and new constitutional symptoms (night sweats, weight loss). His Hgb has paradoxically fallen to 9.8 g/dL. Blood smear shows teardrop cells (dacryocytes), nucleated RBCs, and immature granulocytes. Bone marrow is difficult to aspirate ("dry tap") - biopsy shows dense collagen fibrosis.
What has occurred and what treatment is indicated?
A) He has relapsed PV - increase phlebotomy frequency
B) He has developed post-PV myelofibrosis (spent phase); treat with ruxolitinib
C) He has transformed to CML; start imatinib
D) He has developed iron deficiency from phlebotomy - supplement iron
E) This is a new diagnosis of aplastic anemia
✅ ANSWER: B - Post-PV myelofibrosis; treat with ruxolitinib
Why B is correct:
Post-PV myelofibrosis (spent phase) occurs in 15-20% of PV patients after an average of 10 years. Classic features:
- Paradoxical fall in Hgb (marrow replaced by fibrosis, no longer producing RBCs effectively)
- Massive splenomegaly from extramedullary hematopoiesis compensating for failed marrow
- Leukoerythroblastic blood smear: teardrop cells (dacryocytes - shaped by splenic passage), nucleated RBCs, immature granulocytes
- Dry tap on aspiration + dense marrow fibrosis on biopsy
- Constitutional symptoms (B symptoms)
Treatment: Ruxolitinib (JAK1/2 inhibitor) significantly reduces spleen size and improves symptoms. Allogeneic HSCT should be evaluated in eligible patients as the only potentially curative approach.
Why others are wrong:
- ❌ A: Increasing phlebotomy in a patient with already low Hgb (9.8) would be harmful and is completely inappropriate. The PV has transformed.
- ❌ C: PV does not transform to CML. BCR-ABL1 does not arise from a JAK2-driven clone.
- ❌ D: Iron deficiency causes microcytic anemia, not the full leukoerythroblastic picture with teardrop cells and marrow fibrosis.
- ❌ E: Aplastic anemia has a hypocellular (empty) marrow, not a fibrotic one. There is no prior PV history in aplastic anemia.
Vignette 13 - PV and Budd-Chiari Syndrome
A 42-year-old woman presents with acute-onset right upper quadrant pain, ascites, hepatomegaly, and mild jaundice. No history of liver disease or alcohol use. Doppler ultrasound confirms hepatic vein thrombosis (Budd-Chiari syndrome). Labs: Hgb 17.6 g/dL (women's threshold >16.0), WBC 11,200/µL, platelets 720,000/µL.
What should be the most important next diagnostic step?
A) Liver biopsy to exclude hepatocellular carcinoma
B) Test for JAK2 V617F mutation to evaluate for underlying PV
C) Test for Factor V Leiden to exclude hereditary thrombophilia
D) CT angiography of hepatic veins to confirm the diagnosis
E) Start anticoagulation and discharge without further workup
✅ ANSWER: B - Test for JAK2 V617F (evaluate for underlying PV)
Why B is correct:
Budd-Chiari syndrome (hepatic vein thrombosis) is a classic presenting complication of PV, particularly in young women. PV and other MPNs (ET, PMF) account for 40-50% of all Budd-Chiari syndrome cases. The hallmark is thrombosis of unusual sites (hepatic, portal, mesenteric, splenic veins, cerebral venous sinuses). Even if the Hgb threshold is only marginally elevated, the combination of thrombocytosis + abdominal venous thrombosis should trigger immediate testing for JAK2 V617F. Note: iron deficiency (from occult GI bleeding) can mask the full polycythemic picture, making the Hgb appear less elevated.
Why others are wrong:
- ❌ A: Liver biopsy might be needed eventually, but the priority is identifying the underlying thrombophilic cause, as PV is highly treatable and changes long-term management dramatically.
- ❌ C: Factor V Leiden and other thrombophilias should also be tested, but PV/MPNs are the most common single cause of Budd-Chiari and should be prioritized.
- ❌ D: Doppler has already confirmed the diagnosis. CT angiography adds radiation without changing the urgent diagnostic priority.
- ❌ E: Anticoagulation is important for management but must be paired with identifying the underlying cause. Discharging without PV workup would miss a life-threatening and treatable condition.
Vignette 14 - Aquagenic Pruritus Mechanism
A 57-year-old man with newly diagnosed PV complains bitterly about unbearable itching after swimming or bathing. He describes it as "burning needles" on his skin immediately after contact with warm water. No rash is visible.
What is the most accurate explanation for this symptom, and what treatment can help?
A) Dry skin from dehydration due to elevated RBC mass; use moisturizers
B) Iron deficiency from phlebotomy causing skin changes; supplement iron
C) Histamine release from basophils in the neoplastic clone triggered by temperature change; treat with antihistamines or aspirin
D) Increased IgE from immunologic dysregulation; treat with antihistamines
E) Hyperviscosity causing skin ischemia; phlebotomy alone will resolve it
✅ ANSWER: C - Histamine from basophils; antihistamines or aspirin
Why C is correct:
Aquagenic pruritus (itching triggered specifically by water, particularly warm water) is one of the most characteristic symptoms of PV, occurring in up to 40% of patients. The mechanism is histamine release from the proliferating basophil pool in the neoplastic clone, triggered by temperature change on skin contact with water. No urticarial rash is visible (unlike allergic urticaria). Management includes:
- Antihistamines (H₁ blockers) - first-line for symptoms
- Low-dose aspirin - effective via COX inhibition affecting mast cell/basophil activation
- Ruxolitinib - dramatically reduces pruritus by suppressing JAK-STAT-driven basophil proliferation
- Paroxetine (SSRI) - second-line option (mechanism unclear but clinically effective)
- Best long-term solution: cytoreductive therapy to control the neoplastic clone
Why others are wrong:
- ❌ A: Dry skin occurs in iron deficiency or dehydration but does not cause the characteristic burning water-triggered pattern. Moisturizers would not help.
- ❌ B: Iron deficiency from phlebotomy can cause pruritus (diffuse, not aquagenic). But this patient has newly diagnosed PV and hasn't had phlebotomy-induced iron deficiency yet.
- ❌ D: IgE-mediated allergy causes urticaria (hives). Aquagenic pruritus in PV has no IgE mechanism.
- ❌ E: Phlebotomy reduces viscosity and may help indirectly over time but does not address the immediate histamine-mediated symptom.
Vignette 15 - PV Long-Term Outcome
A 55-year-old man with PV is counselled about his long-term prognosis and transformation risk.
Which of the following best describes the natural history of PV with modern treatment?
A) PV is uniformly fatal within 5 years regardless of treatment
B) Median survival with phlebotomy alone is approximately 10 years; modern therapy (cytoreduction + phlebotomy) significantly reduces thrombotic deaths; ~15-20% transform to post-PV myelofibrosis and 1-2% transform to AML
C) PV is curable with hydroxyurea; no transformation risk
D) Allogeneic HSCT in all patients is necessary to prevent transformation
E) All PV patients will transform to AML within 10 years if untreated
✅ ANSWER: B - Median survival ~10 years; 15-20% post-PV MF; 1-2% AML
Why B is correct:
The natural history of PV with treatment:
- Without treatment: Death from vascular complications within months
- With phlebotomy alone: Median survival ~10 years (Robbins)
- Modern therapy (phlebotomy + cytoreduction + aspirin): 5-year survival >90%; thrombotic death markedly reduced
- Transformation to post-PV myelofibrosis: ~15-20% after average 10 years
- Transformation to AML: ~1-2% (rare but poor prognosis when it occurs; risk is higher with alkylating agent exposure)
- Today, most PV patients die of cardiovascular complications rather than transformation
Why others are wrong:
- ❌ A: PV with treatment has excellent prognosis - 5-year survival exceeds 90% with modern management.
- ❌ C: No treatment is curative for PV (except possibly allogeneic HSCT in selected post-PV MF cases). Hydroxyurea controls the disease but does not eradicate the clone or prevent transformation.
- ❌ D: HSCT in all PV patients would cause unacceptable treatment-related mortality for a disease with >90% 5-year survival on medical management.
- ❌ E: AML transformation occurs in only ~1-2% of PV patients; it is not inevitable. The more common late complication is post-PV myelofibrosis (15-20%).
PART 2: ESSENTIAL THROMBOCYTHEMIA (15 Vignettes)
Vignette 16 - ET Classic Presentation
A 38-year-old woman presents with episodes of burning pain, redness, and warmth of both hands, occurring intermittently and relieved dramatically by aspirin. Platelet count on CBC is 1,180,000/µL with giant platelets on smear. Hgb 12.8 g/dL, WBC 9,200/µL. Peripheral smear shows no immature cells.
What is the most likely diagnosis?
A) Reactive thrombocytosis from iron deficiency
B) Essential thrombocythemia presenting with erythromelalgia
C) CML in chronic phase
D) Primary myelofibrosis
E) Systemic lupus erythematosus with thrombocytosis
✅ ANSWER: B - Essential Thrombocythemia with erythromelalgia
Why B is correct:
Erythromelalgia - episodic burning pain with erythema and warmth of the extremities, dramatically relieved by aspirin - is the pathognomonic microvascular complication of ET (also seen in PV). It is caused by platelet aggregate-mediated occlusion of small arterioles, causing local ischemia. The extreme thrombocytosis (>450,000/µL; here 1,180,000/µL), giant platelets on smear, normal Hgb/WBC, and young female demographics are all consistent with ET. The aspirin-responsiveness is diagnostically meaningful.
Why others are wrong:
- ❌ A (Reactive thrombocytosis): Reactive causes (iron deficiency, inflammation) produce smaller, morphologically normal platelets. Giant platelets suggest a clonal process. Erythromelalgia does not occur in reactive thrombocytosis.
- ❌ C (CML): CML has marked leukocytosis (often >100,000), basophilia, left shift, and BCR-ABL1 positivity. Normal WBC excludes CML here.
- ❌ D (PMF): PMF presents with cytopenias, teardrop cells, leukoerythroblastic picture, and marrow fibrosis - not isolated thrombocytosis in a young woman.
- ❌ E (SLE): SLE causes thrombocytopenia (low platelets), not thrombocytosis. SLE could cause arthralgia, rash, serositis.
Vignette 17 - ET Molecular Workup
A 52-year-old man has a sustained platelet count of 890,000/µL. Reactive causes have been excluded. JAK2 V617F is negative. Bone marrow biopsy shows proliferative megakaryocytes with staghorn/hyperlobulated nuclei and no significant fibrosis.
What is the appropriate next molecular step?
A) BCR-ABL1 FISH - must exclude CML first
B) Test for CALR exon 9 mutation and MPL W515L/K mutation
C) FLT3 and NPM1 mutations for AML
D) Test for TP53 deletion to assess transformation risk
E) No further molecular testing is needed - diagnosis is established by bone marrow alone
✅ ANSWER: B - Test CALR exon 9 and MPL W515 mutations
Why B is correct:
ET molecular frequencies:
- JAK2 V617F: ~50-60%
- CALR (calreticulin) exon 9 frameshift: ~25-35%
- MPL W515L/K: ~5-10%
- Triple-negative (no JAK2/CALR/MPL): ~5-10%
When JAK2 is negative in suspected ET, CALR and MPL must be tested before concluding triple-negative status. CALR mutations are particularly relevant because CALR+ ET has distinct features: higher platelet counts, lower thrombotic risk than JAK2+ ET, more common in younger patients, and generally more favorable prognosis.
Why others are wrong:
- ❌ A: BCR-ABL1 exclusion is part of ET workup (one of the major criteria is "not CML"), but the question asks what to do AFTER JAK2 is negative, and BCR-ABL should already be checked as part of the workup alongside JAK2.
- ❌ C: FLT3/NPM1 are AML markers with no role in ET diagnosis.
- ❌ D: TP53 testing has prognostic value in some MPN/MDS contexts but is not part of primary ET diagnosis criteria.
- ❌ E: Bone marrow biopsy establishes morphology, but molecular clonal markers are required diagnostic criteria by WHO/ICC. A CALR or MPL mutation fulfills a major criterion; missing it would mean incomplete workup.
Vignette 18 - CALR vs JAK2 in ET: Clinical Differences
A haematology trainee is asked to compare JAK2 V617F-positive ET versus CALR-mutated ET.
Which of the following correctly contrasts JAK2+ ET with CALR+ ET?
A) CALR+ ET has higher thrombotic risk than JAK2+ ET
B) JAK2+ ET has higher Hgb levels, higher WBC, higher thrombotic risk; CALR+ ET has higher platelet counts and lower thrombotic risk
C) Both mutations confer identical clinical risk profiles
D) CALR+ ET is always more aggressive and requires earlier cytoreduction
E) JAK2 mutations are found only in women with ET; CALR mutations only in men
✅ ANSWER: B - JAK2+ = higher Hgb/WBC/thrombotic risk; CALR+ = higher platelets, lower thrombotic risk
Why B is correct:
| Feature | JAK2 V617F+ ET | CALR+ ET |
|---|---|---|
| Thrombotic risk | Higher | Lower |
| Hgb | Often higher (may overlap with PV) | Normal |
| WBC | Often higher | Normal/lower |
| Platelet count | Moderate elevation | Often extremely high |
| Age | Older | Younger |
| Transformation to MF | Higher rate | Lower rate |
| Prognosis | Slightly worse | More favorable |
JAK2 V617F activates the EPO/TPO/G-CSF receptor pathways more broadly (explaining panmyeloid features), while CALR mutations specifically activate the thrombopoietin receptor (explaining extreme platelet-selective proliferation).
Why others are wrong:
- ❌ A: The opposite is true - JAK2+ ET carries higher thrombotic risk.
- ❌ C: The mutations have distinct clinical profiles with meaningful differences in risk stratification.
- ❌ D: CALR+ ET generally has a more favorable prognosis than JAK2+ ET.
- ❌ E: No sex-specific mutation distribution exists in ET.
Vignette 19 - ET and Extreme Thrombocytosis: Paradoxical Bleeding
A 63-year-old man with ET has a platelet count of 2,450,000/µL. He is due for a right hemicolectomy for colon cancer. During preoperative assessment, he reports easy bruising. Ristocetin cofactor activity returns at 18% (normal >50%). VWF multimer analysis shows absence of high-molecular-weight multimers.
What is the mechanism of his bleeding risk, and what must be done before surgery?
A) Thrombocytopenia-induced bleeding; transfuse platelets preoperatively
B) Acquired von Willebrand disease from platelet adsorption of large VWF multimers; reduce platelet count with cytoreduction before surgery
C) Antiplatelet antibody causing platelet dysfunction; give IVIG
D) Vitamin K deficiency; give vitamin K preoperatively
E) DIC from the underlying malignancy; treat with FFP and cryoprecipitate
✅ ANSWER: B - Acquired VWD; reduce platelets before surgery
Why B is correct:
When platelet counts exceed ~1,000,000-1,500,000/µL in ET, the abnormally high density of platelets adsorb and remove the largest, most hemostatically effective von Willebrand factor multimers from circulation. This creates acquired von Willebrand disease (Type 2A-like pattern):
- Low ristocetin cofactor activity (<30% is diagnostic)
- Absent high-molecular-weight VWF multimers on analysis
- Paradoxically high platelets but bleeding tendency from loss of functional VWF
Management: Cytoreductive therapy (hydroxyurea, platelet apheresis in emergency) to reduce platelet count to <1,000,000/µL restores VWF multimers and normalizes hemostasis. Surgery should be deferred until platelet counts are safely reduced and ristocetin cofactor activity normalizes.
Why others are wrong:
- ❌ A: Platelets are massively elevated, not low. Transfusing more platelets worsens aVWD.
- ❌ C: IVIG treats immune thrombocytopenia (ITP, low platelets from immune destruction) - not applicable here.
- ❌ D: Vitamin K deficiency affects factors II, VII, IX, X. This patient's picture is specifically VWF-related.
- ❌ E: DIC involves consumption coagulopathy (low fibrinogen, elevated D-dimer, thrombocytopenia). The platelet count here is extremely high, excluding DIC.
Vignette 20 - ET Diagnosis: Exclusion of Reactive Causes
A 44-year-old woman has persistent thrombocytosis (platelet count 620,000/µL on three consecutive CBCs over 6 months). She had a prior splenectomy for trauma 2 years ago. CRP is mildly elevated at 18 mg/L. Iron studies show ferritin 8 ng/mL (low) and low serum iron.
What should be done before diagnosing ET?
A) Immediately send JAK2/CALR/MPL and perform bone marrow biopsy - this is ET
B) Address reversible causes (treat iron deficiency, investigate elevated CRP for underlying inflammation) and reassess platelet count; diagnose ET only if thrombocytosis persists after correction
C) Thrombocytosis post-splenectomy is always permanent; start hydroxyurea
D) Diagnose ET based on platelet count alone (>450,000/µL)
E) The elevated CRP confirms ET - inflammatory markers are always elevated in ET
✅ ANSWER: B - Treat reversible causes first; then reassess
Why B is correct:
Reactive (secondary) thrombocytosis is far more common than ET and has several causes:
- Post-splenectomy (no splenic platelet sequestration)
- Iron deficiency (a well-recognized cause of thrombocytosis)
- Inflammation/infection (elevated CRP = inflammatory state driving reactive megakaryocytopoiesis via IL-6 → thrombopoietin)
The WHO/ICC ET diagnostic criteria explicitly require:
- Sustained platelet count ≥450,000/µL
- NOT meeting criteria for PV, PMF, MDS, or CML
- Reactive causes excluded
Before proceeding to molecular and marrow testing, treat the iron deficiency and investigate the elevated CRP. If thrombocytosis resolves → reactive cause confirmed. If it persists → proceed with molecular workup.
Why others are wrong:
- ❌ A: Jumping to ET workup without addressing obvious reactive causes leads to overdiagnosis and potential harm from unnecessary cytoreductive therapy.
- ❌ C: Post-splenectomy thrombocytosis is often transient and decreases over months. It is NOT always permanent and does not automatically mean ET.
- ❌ D: Platelet count alone is insufficient. The WHO/ICC requires multiple criteria including exclusion of reactive causes and bone marrow/molecular confirmation.
- ❌ E: CRP elevation suggests inflammation (a reactive cause) - not ET specifically. ET itself does not typically elevate CRP.
Vignette 21 - ET Thrombosis Risk Score
A 67-year-old man with ET (JAK2 V617F+) had a stroke 2 years ago. Platelet count is 980,000/µL. He has diabetes and hypertension.
Using the IPSET-thrombosis score, how is this patient classified?
A) Low-risk; aspirin alone is appropriate
B) Intermediate-risk; aspirin ± cytoreduction
C) Very high-risk; requires cytoreduction (hydroxyurea) + aspirin
D) Cannot be classified without CALR mutation status
E) High-risk by age alone; aspirin only is sufficient
✅ ANSWER: C - Very high-risk; hydroxyurea + aspirin
Why C is correct:
The revised IPSET-thrombosis classification:
| Category | Criteria |
|---|---|
| Very High-risk | Prior thrombosis OR age >60 + JAK2 V617F |
| High-risk | Age >60 OR JAK2 V617F (without prior thrombosis) |
| Intermediate-risk | Age ≤60 without prior thrombosis, JAK2 V617F+ |
| Low-risk | Age ≤60, JAK2 V617F-, no thrombosis |
This patient: age 67 (>60) + JAK2 V617F + prior stroke = Very High-risk → cytoreductive therapy with hydroxyurea (first-line) + low-dose aspirin is mandatory. Cardiovascular risk factors (diabetes, hypertension) add further risk.
Why others are wrong:
- ❌ A: Low-risk applies to young (<60), JAK2-negative, thrombosis-free patients. This patient is the opposite.
- ❌ B: Intermediate-risk management would under-treat this very high-risk patient.
- ❌ D: Thrombosis risk scoring uses age, JAK2 status, and prior thrombosis - CALR status is not a component of IPSET scoring (though CALR+ generally has lower risk).
- ❌ E: Age alone places him in high-risk, but the additional prior thrombosis and JAK2+ status makes him very high-risk requiring cytoreduction, not aspirin alone.
Vignette 22 - ET Bone Marrow Morphology
A 58-year-old woman with ET undergoes bone marrow biopsy. The pathologist reports enlarged megakaryocytes with hyperlobulated "staghorn" or "antler" nuclei clustering in loose groups. Granulocyte and erythroid series appear normal. Reticulin fibrosis is minimal.
What does this morphology confirm, and how does it differ from PMF megakaryocytes?
A) The staghorn megakaryocytes confirm PMF; start ruxolitinib
B) The mature, hyperlobulated megakaryocytes with minimal fibrosis confirm ET; PMF megakaryocytes are smaller with hypolobulated "cloud-like" nuclei and marked fibrosis
C) These are normal megakaryocytes; no diagnosis can be made from morphology alone
D) This pattern is identical to CML megakaryocytes
E) Staghorn megakaryocytes indicate AML-M7 (megakaryoblastic)
✅ ANSWER: B - Confirms ET; PMF has small hypolobulated megakaryocytes with fibrosis
Why B is correct:
Bone marrow megakaryocyte morphology is a critical differentiating feature:
| Feature | ET | PMF |
|---|---|---|
| Size | Large, mature | Variable; often small/immature |
| Nuclei | Hyperlobulated "staghorn/antler" | Hypolobulated, "cloud-like," immature |
| Clustering | Loose groups | Tight clusters around sinuses/trabeculae |
| Fibrosis | Absent or minimal | Prominent reticulin/collagen fibrosis |
| Overall cellularity | Normal to mildly increased | Hypercellular (early) → fibrotic (late) |
This distinction is one of the key reasons bone marrow biopsy with expert morphological review is required to distinguish ET from prefibrotic PMF (which can mimic ET clinically).
Why others are wrong:
- ❌ A: PMF megakaryocytes are the opposite - small with cloud-like nuclei and associated with dense fibrosis. This biopsy shows ET features.
- ❌ C: Megakaryocyte morphology is a major criterion in the WHO/ICC diagnostic classification of MPNs.
- ❌ D: CML megakaryocytes are small and hypolobulated (similar to PMF), not the staghorn pattern.
- ❌ E: AML-M7 (acute megakaryoblastic leukemia) has megakaryoblasts (≥20% blasts of megakaryocytic lineage), CD41/CD61 positivity - a completely different entity.
Vignette 23 - ET and Pregnancy Complications
A 29-year-old woman with JAK2 V617F+ ET (platelet count 1,100,000/µL) has had two first-trimester miscarriages and one stillbirth at 24 weeks. She is now 6 weeks pregnant again.
What is the optimal management strategy for this high-risk pregnancy?
A) No treatment is needed; miscarriages in ET are coincidental
B) Low-dose aspirin + low-molecular-weight heparin (LMWH) + interferon-alpha for cytoreduction; avoid hydroxyurea
C) Hydroxyurea + warfarin throughout pregnancy
D) Therapeutic abortion is recommended in all ET pregnancies
E) Platelet transfusion to normalize platelet count before delivery
✅ ANSWER: B - Aspirin + LMWH + interferon-alpha; avoid hydroxyurea
Why B is correct:
ET in pregnancy carries significant risk:
- First-trimester loss: Placental microvascular thrombosis from platelet aggregates
- Later complications: Placental infarction, abruption, IUGR, stillbirth
- Maternal: DVT, PE, thrombotic events
This patient with prior pregnancy losses is high-risk and requires:
- Low-dose aspirin (reduces placental thrombosis)
- LMWH (subcutaneous heparin - safe in pregnancy, does not cross placenta) - for anticoagulation given prior losses
- Interferon-alpha (if cytoreduction needed - safe, does not cross placenta)
- Hydroxyurea is ABSOLUTELY CONTRAINDICATED in pregnancy (teratogenic)
- Anagrelide is also contraindicated in pregnancy
Why others are wrong:
- ❌ A: ET is a proven cause of recurrent pregnancy loss; ignoring it is negligent.
- ❌ C: Hydroxyurea = teratogenic (contraindicated). Warfarin crosses the placenta and causes warfarin embryopathy (nasal hypoplasia, stippled epiphyses, CNS anomalies) - contraindicated in first trimester.
- ❌ D: Therapeutic abortion is not routinely recommended. With proper management, successful pregnancy is achievable.
- ❌ E: Platelet transfusion would worsen thrombosis in a patient with already extreme thrombocytosis and dysfunction.
Vignette 24 - ET: First-Line Cytoreductive Agent
A 70-year-old woman with ET (JAK2 V617F+) had a DVT 6 months ago. Platelet count is 1,350,000/µL. She has no other comorbidities.
What is the first-line cytoreductive agent for this high-risk ET patient?
A) Anagrelide
B) Busulfan
C) Hydroxyurea (hydroxycarbamide) + low-dose aspirin
D) Ruxolitinib
E) Interferon-alpha
✅ ANSWER: C - Hydroxyurea + low-dose aspirin
Why C is correct:
Hydroxyurea (hydroxycarbamide) is the established first-line cytoreductive agent for high-risk ET based on the landmark PT-1 trial, which showed hydroxyurea + aspirin was superior to anagrelide + aspirin in reducing arterial thrombosis, bleeding, and transformation to myelofibrosis. Hydroxyurea:
- Inhibits ribonucleotide reductase → reduces DNA synthesis in rapidly dividing cells
- Effectively reduces platelet count to target (<400,000-600,000/µL)
- Well-tolerated in most patients
- Target: platelet count <400,000/µL ideally; at minimum <600,000/µL
Why others are wrong:
- ❌ A (Anagrelide): Anagrelide is a second-line option (when hydroxyurea fails or is intolerant). It selectively inhibits megakaryocyte differentiation. The PT-1 trial showed it was inferior to hydroxyurea as first-line.
- ❌ B (Busulfan): Busulfan is an alkylating agent with leukemogenic potential. It is used in elderly patients or when other agents fail - not first-line.
- ❌ D (Ruxolitinib): Ruxolitinib is used in PV refractory to HU and in myelofibrosis; it is not standard first-line for ET.
- ❌ E (Interferon-alpha): IFN-α is preferred in young patients (<40) and pregnant women. For a 70-year-old post-thrombosis patient, hydroxyurea is preferred as it is better tolerated and has robust trial data.
Vignette 25 - ET vs Prefibrotic PMF
A 55-year-old woman presents with thrombocytosis (platelet count 820,000/µL). She has mild splenomegaly. Bone marrow biopsy shows megakaryocytic proliferation but the megakaryocytes are small with hypolobulated "cloud-like" nuclei, clustering tightly around sinuses. Mild reticulin fibrosis (MF-1). No overt collagen fibrosis.
What is the most accurate diagnosis, and why does it matter?
A) Essential thrombocythemia - staging and prognosis are the same as prefibrotic PMF
B) Prefibrotic PMF - clinically mimics ET but has a worse prognosis with higher rate of fibrotic transformation and lower overall survival; requires different management
C) Polycythemia vera with masked polycythemia - treat with phlebotomy
D) Reactive thrombocytosis - no treatment needed
E) CML - BCR-ABL1 testing is required
✅ ANSWER: B - Prefibrotic PMF; worse prognosis than ET
Why B is correct:
Prefibrotic (early) PMF is a critical diagnostic distinction from ET because it:
- Clinically resembles ET (thrombocytosis, mild splenomegaly, few symptoms)
- Histologically differs: Small, immature megakaryocytes with cloud-like/hypolobulated nuclei, tight perisinusoidal clustering, mild reticulin fibrosis (MF-0 to MF-1) - the opposite of ET's large staghorn megakaryocytes
- Has significantly worse outcomes: Higher rate of progression to overt fibrotic PMF (25-30% vs <5% in ET), lower overall survival, higher transformation to AML
- Management differs: More aggressive monitoring; earlier consideration of cytoreduction and HSCT evaluation
This distinction requires expert bone marrow pathological review and is why bone marrow biopsy is mandatory in all suspected MPNs.
Why others are wrong:
- ❌ A: Prognosis and outcomes are NOT the same. Prefibrotic PMF has median survival of 15 years vs >20 years in ET. The distinction matters significantly for counseling and follow-up intensity.
- ❌ C: Nothing here suggests masked PV (iron deficiency hiding true erythrocytosis). EPO level and JAK2 status would clarify.
- ❌ D: Giant/abnormal megakaryocytes with clustering on biopsy are never "reactive."
- ❌ E: CML features are absent (no leukocytosis, no basophilia, no BCR-ABL1 indication from this picture).
Vignette 26 - Anagrelide Mechanism and Use
A 35-year-old woman with ET is intolerant of hydroxyurea (severe oral ulcers). Her physician considers switching to anagrelide.
What is the mechanism of action of anagrelide?
A) Inhibits ribonucleotide reductase, reducing DNA synthesis in all dividing cells
B) Inhibits JAK1/2 signaling downstream of the thrombopoietin receptor
C) Selectively inhibits megakaryocyte maturation and differentiation by inhibiting phosphodiesterase III, reducing platelet production
D) Directly destroys platelets via complement activation
E) Blocks the thrombopoietin receptor (MPL), preventing megakaryocyte proliferation
✅ ANSWER: C - Inhibits PDE-III → impairs megakaryocyte maturation → fewer platelets
Why C is correct:
Anagrelide is a selective PDE-III (phosphodiesterase type III) inhibitor that specifically disrupts megakaryocyte maturation and differentiation, reducing platelet production without major effects on other cell lines. Key points:
- Selective for platelets (unlike hydroxyurea which affects all rapidly dividing cells)
- Advantages: Useful when HU-intolerant, no leukemogenic potential
- Disadvantages: Cardiac side effects (palpitations, fluid retention, arrhythmia - from PDE-III inhibition in cardiac muscle), headaches, diarrhea
- Contraindicated in cardiac disease
- Not recommended in pregnancy (limited safety data)
- Second-line in most guidelines
Why others are wrong:
- ❌ A: This describes hydroxyurea's mechanism (ribonucleotide reductase inhibition).
- ❌ B: JAK1/2 inhibition is the mechanism of ruxolitinib.
- ❌ D: No cytotoxic/complement-mediated platelet destruction mechanism is involved.
- ❌ E: MPL receptor blockade is not the mechanism; anagrelide works downstream at the intracellular signaling level (PDE-III inhibition).
Vignette 27 - ET: Venous Sinus Thrombosis
A 32-year-old woman presents with a 2-week history of progressive headache, papilledema on fundoscopy, and diplopia. MRI with MR venography confirms cerebral venous sinus thrombosis (CVST). She is not on oral contraceptives. Platelet count is 960,000/µL.
What underlying diagnosis should be urgently investigated?
A) Factor V Leiden only
B) Antiphospholipid syndrome only
C) Essential thrombocythemia (or PV) - send JAK2/CALR/MPL; this is a classic unusual-site thrombosis
D) Protein C and S deficiency only
E) Dehydration-related CVST; rehydrate and discharge
✅ ANSWER: C - ET/PV - unusual site thrombosis; send JAK2/CALR/MPL
Why C is correct:
Cerebral venous sinus thrombosis (CVST) is a classic "unusual site thrombosis" that can be the presenting manifestation of ET or PV, particularly in young women without typical risk factors (OCP, dehydration, infection). The thrombocytosis of 960,000/µL is a major red flag. MPNs (ET/PV) are strongly associated with:
- Splanchnic vein thrombosis (portal, hepatic, mesenteric - Budd-Chiari)
- Cerebral venous sinus thrombosis
- Retinal vein/artery thrombosis
In any young patient with unusual site thrombosis + abnormal CBC, MPN must be excluded urgently by JAK2 V617F testing (positive in >97% of PV and ~50-60% of ET), followed by CALR/MPL if negative.
Why others are wrong:
- ❌ A, B, D: All thrombophilias should be tested, but the thrombocytosis of 960,000 strongly suggests an MPN as the primary etiology. Inherited thrombophilias (Factor V Leiden, protein C/S, antiphospholipid syndrome) do not explain thrombocytosis.
- ❌ E: Dehydration can contribute to CVST but does not explain extreme thrombocytosis. Discharging without investigating the thrombocytosis is dangerous.
Vignette 28 - ET: Post-ET Myelofibrosis
A 68-year-old man with a 15-year history of ET (CALR mutation) presents with progressive splenomegaly, increasing fatigue, and new constitutional symptoms. His platelet count has paradoxically fallen to 180,000/µL. Blood smear shows teardrop cells and nucleated RBCs. Bone marrow biopsy shows grade 3 reticulin fibrosis.
What has occurred and what treatment should be considered?
A) ET has spontaneously resolved - no treatment needed
B) He has developed post-ET myelofibrosis; evaluate for ruxolitinib and allogeneic HSCT in eligible patients
C) He now has aplastic anemia from hydroxyurea toxicity
D) His CALR mutation has converted to JAK2 V617F
E) This represents reactive fibrosis from a viral infection
✅ ANSWER: B - Post-ET myelofibrosis; ruxolitinib ± HSCT
Why B is correct:
Post-ET myelofibrosis occurs in approximately 5% of ET patients over 10+ years (less than post-PV MF frequency of ~15-20%). Features mirror post-PV MF:
- Falling platelet count (marrow fibrosis replaces megakaryopoiesis)
- Splenomegaly (extramedullary hematopoiesis)
- Constitutional symptoms (B symptoms)
- Leukoerythroblastic smear (teardrop cells, NRBCs, immature WBCs)
- Marrow fibrosis on biopsy (reticulin grade 2-3 or collagen fibrosis)
Management: Ruxolitinib for symptom/spleen control; allogeneic HSCT for eligible patients as the only potential cure (HSCT decision guided by DIPSS/DIPSS-Plus scoring).
Why others are wrong:
- ❌ A: This is disease transformation, not spontaneous resolution. The fibrotic marrow means worsening disease.
- ❌ C: Aplastic anemia has hypocellular (empty) marrow, not fibrotic marrow. The ET history makes MF transformation far more likely.
- ❌ D: Clonal evolution (acquiring new mutations) can occur but mutations don't "convert" from CALR to JAK2. Additional mutations in ASXL1, EZH2, or TP53 drive transformation to MF.
- ❌ E: Grade 3 reticulin fibrosis in the context of a known MPN is clonal, not reactive.
Vignette 29 - ET: Aspirin and Paradoxical Thrombosis Risk
A 46-year-old woman with JAK2 V617F+ ET (platelet count 1,800,000/µL) is about to be started on low-dose aspirin by her internist. A hematologist is asked to review.
Why should caution be exercised before starting aspirin in this patient?
A) Aspirin has no role in ET; it is contraindicated
B) At platelet counts >1,000,000-1,500,000/µL, acquired VWD from loss of large VWF multimers means aspirin may worsen bleeding risk; ristocetin cofactor activity should be checked first
C) Aspirin should only be used in JAK2-negative ET
D) Aspirin will cause thrombocytopenia in ET
E) Aspirin interacts with hydroxyurea and should not be combined
✅ ANSWER: B - Check ristocetin cofactor activity; aspirin may worsen bleeding in aVWD
Why B is correct:
This is a nuanced and important clinical point: While aspirin is generally beneficial in ET (reduces microvascular thrombosis), at extreme thrombocytosis (>1,000,000-1,500,000/µL), acquired von Willebrand disease (aVWD) is present due to adsorption of large VWF multimers by excess platelets. In this context:
- The patient already has a hemostatic defect from aVWD
- Adding aspirin (which further impairs platelet function via COX-1 inhibition) may potentiate bleeding risk
- Check ristocetin cofactor activity (<30% = significant aVWD) before starting aspirin
- If aVWD is confirmed, reduce platelet count first with cytoreduction (hydroxyurea), then reassess for aspirin use
Why others are wrong:
- ❌ A: Aspirin is not contraindicated in ET broadly - it is recommended for most ET patients. The concern is specific to extreme thrombocytosis with confirmed aVWD.
- ❌ C: Aspirin is used in both JAK2+ and JAK2- ET based on symptoms and risk stratification, not mutation status alone.
- ❌ D: Aspirin does not cause thrombocytopenia.
- ❌ E: Aspirin and hydroxyurea are commonly used together in high-risk ET - there is no dangerous interaction.
Vignette 30 - Integrated ET Case
A 54-year-old woman presents with an incidentally noted platelet count of 750,000/µL. She has mild headaches and one episode of transient right arm numbness last month (resolved in 30 minutes). She has no prior thrombosis history. Physical exam: mild splenomegaly. CBC: Hgb 13.4 g/dL, WBC 10,200/µL. JAK2 V617F: positive. BCR-ABL1: negative. Bone marrow biopsy: large hyperlobulated megakaryocytes, minimal reticulin fibrosis.
Step 1: What is the diagnosis?
Step 2: What is her risk category?
Step 3: What is the appropriate management?
A) ET, low-risk, observation only
B) ET, very high-risk (JAK2+ + age >40 + TIA = prior thrombotic event), hydroxyurea + aspirin
C) PV, high-risk, phlebotomy + hydroxyurea
D) PMF, intermediate-2, ruxolitinib
E) ET, intermediate-risk, aspirin alone
✅ ANSWER: B - ET, Very High-Risk, Hydroxyurea + Aspirin
Why B is correct:
Step 1 - Diagnosis: ET confirmed by:
- Platelet count ≥450,000/µL (750,000) ✓
- JAK2 V617F positive ✓
- BCR-ABL1 negative (excludes CML) ✓
- Bone marrow: large hyperlobulated megakaryocytes, minimal fibrosis ✓ (ET morphology)
- Normal Hgb (excludes PV) ✓
Step 2 - Risk Category: The TIA last month = prior thrombotic event + JAK2 V617F+ = Very High-risk by revised IPSET-thrombosis. (Note: some classifications consider TIA as prior cerebrovascular event, qualifying as prior thrombosis.)
Step 3 - Management:
- Hydroxyurea (first-line cytoreductive therapy for high/very high-risk ET)
- Low-dose aspirin (reduces microvascular and thrombotic events)
- Target platelet count <400,000-600,000/µL
- Monitor CBC every 3 months; bone marrow reassessment if symptoms change
Why others are wrong:
- ❌ A: Low-risk management (observation) is completely inappropriate given a prior TIA and JAK2+ status. This patient requires cytoreduction.
- ❌ C: PV requires elevated Hgb (>16.0 in women) and/or elevated Hct (>48%). Her Hgb is 13.4 g/dL - normal. This is ET.
- ❌ D: PMF features (teardrop cells, marrow fibrosis, cytopenias, leukoerythroblastosis) are absent. The marrow shows ET morphology.
- ❌ E: Aspirin alone is insufficient for very high-risk ET with prior thrombosis. Cytoreduction is mandatory.
Summary Reference Table
| # | Topic | Key Point |
|---|---|---|
| 1 | PV classic presentation | Aquagenic pruritus + low EPO + panmyelosis |
| 2 | PV diagnostic criteria | 2 major + 1 minor = diagnosis; BM not always mandatory |
| 3 | PV pathogenesis | JAK2 V617F → EPO-independent → low EPO |
| 4 | PV vs relative polycythemia | Normal EPO + normal WBC/plt + JAK2-negative = relative |
| 5 | PV and TIA/stroke | Hyperviscosity thrombosis; high-risk trigger |
| 6 | PV risk stratification | Age ≥60 OR prior thrombosis = high-risk → HU |
| 7 | Phlebotomy target | Hct <45% (CYTO-PV trial evidence) |
| 8 | Hydroxyurea toxicity | Leg/oral ulcers + macrocytosis → ruxolitinib/IFN-α |
| 9 | PV and gout | High cell turnover → uric acid → allopurinol |
| 10 | PV and peptic ulcer | Basophil histamine → H₂ receptor → gastric acid |
| 11 | PV in pregnancy | IFN-α safe; HU contraindicated; aspirin allowed |
| 12 | Post-PV myelofibrosis | Paradoxical Hgb fall + teardrop cells + marrow fibrosis |
| 13 | PV and Budd-Chiari | JAK2 V617F test in any young patient with hepatic vein thrombosis |
| 14 | Aquagenic pruritus | Histamine from basophils; antihistamines + aspirin |
| 15 | PV long-term prognosis | 15-20% → post-PV MF; 1-2% → AML |
| 16 | ET classic (erythromelalgia) | Aspirin-responsive platelet microvascular occlusion |
| 17 | ET molecular workup | JAK2 negative → test CALR exon 9 + MPL W515 |
| 18 | JAK2 vs CALR ET | JAK2+ = higher thrombotic risk; CALR+ = higher platelets, lower risk |
| 19 | Extreme thrombocytosis bleeding | Acquired VWD from VWF multimer adsorption; reduce PLT before surgery |
| 20 | ET: exclusion of reactive causes | Treat iron deficiency/inflammation first; then reassess |
| 21 | ET risk stratification | IPSET: age >60 + JAK2+ + prior thrombosis = very high-risk |
| 22 | ET bone marrow morphology | Staghorn megakaryocytes vs PMF's cloud-like small megakaryocytes |
| 23 | ET in pregnancy | Aspirin + LMWH + IFN-α; HU absolutely contraindicated |
| 24 | ET first-line cytoreduction | Hydroxyurea (PT-1 trial) > anagrelide as first-line |
| 25 | ET vs prefibrotic PMF | Morphology distinguishes; prefibrotic PMF has worse prognosis |
| 26 | Anagrelide mechanism | PDE-III inhibitor → impairs megakaryocyte maturation |
| 27 | ET: unusual site thrombosis | CVST in young women → check JAK2/CALR/MPL |
| 28 | Post-ET myelofibrosis | 5% over 15 years; ruxolitinib ± HSCT |
| 29 | Aspirin caution in extreme thrombocytosis | aVWD at PLT >1.5M; check ristocetin cofactor first |
| 30 | Integrated ET case | Diagnosis + risk + treatment in one case |
Sources: Robbins Pathologic Basis of Disease, Goldman-Cecil Medicine, Harrison's Principles of Internal Medicine 22E, Henry's Clinical Diagnosis and Management by Laboratory Methods, Washington Manual of Medical Therapeutics
This is a shared conversation. Sign in to Orris to start your own chat.