Testicular feminisation syndrome
Reading File
Finding Sources
Searching PubMed
androgen insensitivity syndrome
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Here is a detailed, authoritative overview of Testicular Feminisation Syndrome (now more precisely called Complete Androgen Insensitivity Syndrome, CAIS):
Testicular Feminisation Syndrome (Complete Androgen Insensitivity Syndrome)
Terminology
The older eponym "testicular feminisation syndrome" has been replaced in modern clinical practice by Complete Androgen Insensitivity Syndrome (CAIS). It falls under the broader category of Disorders of Sex Development (DSD), specifically 46,XY DSD.
Genetics and Pathophysiology
- Karyotype: 46,XY
- Inheritance: X-linked recessive (the androgen receptor gene, AR, is on the X chromosome at Xq11-q12)
- Incidence: ~1 in 10,000-20,000 live births
- Molecular defect: Loss-of-function mutations in the AR gene - ranging from complete gene deletion to point mutations affecting the androgen-binding or DNA-binding domain of the androgen receptor protein
The core mechanism:
The androgen receptor (AR) normally forms a complex with testosterone and dihydrotestosterone (DHT). This complex then acts as a transcription factor to activate genes responsible for male phenotypic differentiation. In CAIS, the AR is absent or non-functional, so:
- Testosterone cannot act on target tissues - the wolffian ducts do not develop into the male internal genitalia (epididymis, vas deferens, seminal vesicles)
- The external genitalia do not masculinise - the genital tubercle, labioscrotal folds, and urethral folds all develop along the female "default" pathway
- The testes still secrete Anti-Mullerian Hormone (AMH) normally - so Mullerian duct structures (uterus, fallopian tubes, upper vagina) are suppressed and absent
This explains the paradoxical result: no female internal organs AND no male internal/external organs - except for the short blind-ending vagina (derived from the urogenital sinus, which forms independently of androgens).
Clinical Features
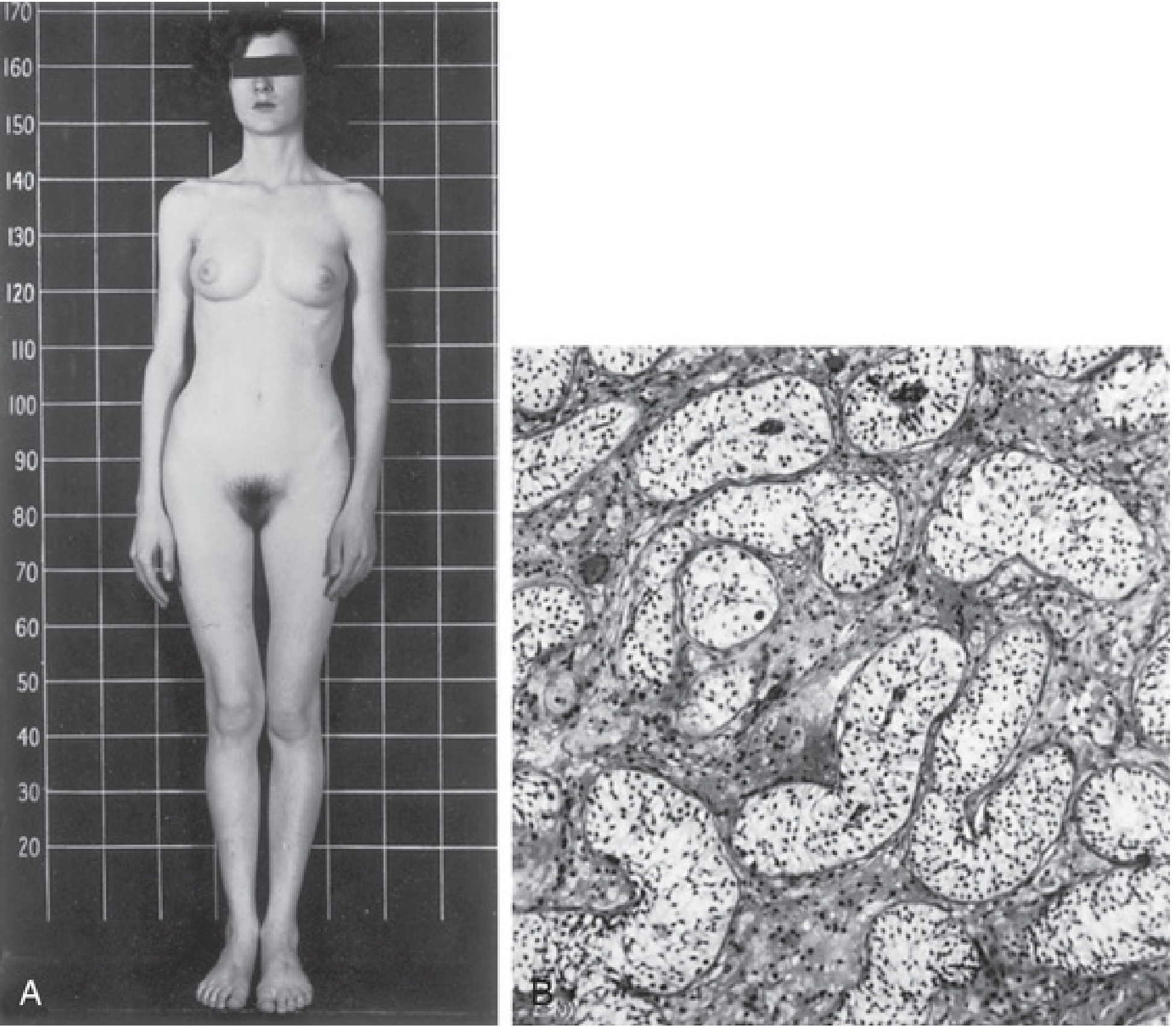
Fig. A: Classic female phenotype in CAIS. Fig. B: Testicular histology - seminiferous tubules lined by Sertoli cells, no germ cells, hypoplastic Leydig cells. (The Developing Human, 9th ed.)
| Feature | Details |
|---|---|
| External genitalia | Unambiguously female - normal clitoris, labia majora (may be hypoplastic), labia minora |
| Vagina | Short, blind-ending pouch; no cervix |
| Uterus / tubes | Absent (suppressed by AMH in utero) |
| Gonads | Testes - located in abdomen, inguinal canal, or labia majora |
| Breast development | Normal (often above average) - from peripheral aromatisation of testosterone to oestradiol |
| Pubic/axillary hair | Sparse to absent (requires functional AR) |
| Menstruation | Primary amenorrhoea - the most common presenting complaint |
| Height | Often above average female height |
| Gender identity | Female - patients identify as female |
Hormonal Profile
| Hormone | Level | Reason |
|---|---|---|
| Testosterone | Mildly elevated (male range) | Normal testicular production, but not acting peripherally |
| LH | Elevated | Absent negative feedback from androgens on the hypothalamus/pituitary |
| FSH | Normal or slightly elevated | |
| Oestradiol | Present | Peripheral aromatisation of testosterone by adipose tissue and Sertoli cells |
The high LH drives the testes to produce increased testosterone, which aromatises to oestradiol - this is what drives breast development at puberty.
Presentations by Age
- Neonate/infant: Inguinal hernia containing a testis in a phenotypic female (CAIS accounts for ~1% of all girls undergoing inguinal hernia repair - this is an important clinical clue)
- Adolescent: Primary amenorrhoea with normal breast development but absent pubic/axillary hair
- Adult: Infertility workup or incidental finding
Diagnosis
- Chromosomal analysis: 46,XY karyotype in a phenotypic female
- Pelvic ultrasound/MRI: Absent uterus and ovaries; testes may be visualised in the abdomen or inguinal canal
- Hormonal studies: Elevated LH and testosterone; oestradiol present
- Androgen-binding studies on genital skin fibroblasts: Absent or defective androgen receptor binding
- Genetic testing: Mutation analysis of the AR gene confirms the diagnosis
Management
Gonadectomy (Gonadal Removal)
The retained testes carry a significant cancer risk:
- ~10% risk of malignant germ cell tumour (most commonly seminoma) after puberty, rising to ~33% by age 50 in some series
- A 2026 systematic review on gonadal malignancy screening in AIS (PMID 41102125) and a 2026 systematic review on testicular cancer in intersex individuals (PMID 41508675) both confirm this elevated risk, reinforcing current guidelines
- Current recommendation: Gonadectomy is advised, preferably after puberty (to allow spontaneous breast development from oestradiol), though some centres remove gonads at diagnosis with subsequent oestrogen replacement
Hormone Replacement Therapy
- Following gonadectomy, oestrogen replacement therapy is required to maintain secondary sex characteristics, prevent osteoporosis, and provide cardiovascular protection
Vaginal Management
- The vaginal pouch may be too short for comfortable sexual intercourse
- First-line: Progressive self-dilation (Frank's technique)
- Surgical options: Vaginoplasty (using skin grafts or bowel segment) if dilation fails
Psychological Support
- Sensitive, ongoing counselling regarding diagnosis, fertility, and gender identity
- Patients should be informed they cannot bear children (no uterus or ovaries)
Comparison: Complete vs. Partial AIS
| Feature | Complete AIS (CAIS) | Partial AIS (PAIS) |
|---|---|---|
| External genitalia | Unambiguously female | Ambiguous (enlarged clitoris, partial fusion) |
| Pubic/axillary hair | Absent | May be present |
| Presentation | Primary amenorrhoea, inguinal hernia | Ambiguous genitalia at birth |
| AR defect | Complete loss of function | Partial - point mutations |
Key Summary Points
- CAIS is caused by X-linked loss-of-function mutations in the androgen receptor (AR) gene
- 46,XY karyotype with female phenotype, absent uterus/tubes, short blind vagina, absent pubic/axillary hair, normal breasts
- Testes present (often inguinal) - AMH is secreted normally, suppressing Mullerian structures
- Oestradiol from peripheral aromatisation of testosterone drives breast development
- Primary amenorrhoea is the classic adolescent presentation
- Gonadectomy recommended due to malignancy risk (~10%), followed by oestrogen HRT
- Inheritance is X-linked recessive; the mother is a carrier
Sources: Smith and Tanagho's General Urology (19th ed.), The Developing Human Clinically Oriented Embryology, Costanzo Physiology (7th ed.), Thompson & Thompson Genetics and Genomics in Medicine (9th ed.), Harrison's Principles of Internal Medicine (22nd ed., 2025)
This is a shared conversation. Sign in to Orris to start your own chat.