DISCUSS MECHANISM OF INVASION AND METASTASIS.
Reading File
Finding Sources
Searching PubMed
"tumor invasion" AND "metastasis" AND "mechanisms"
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the material needed for a thorough, well-illustrated answer. Let me compile the comprehensive discussion:
Mechanism of Invasion and Metastasis
Invasion and metastasis are the major causes of cancer-related morbidity and mortality and together represent one of the most defining hallmarks of malignancy. The process involves a complex, multi-step cascade of interactions between cancer cells, stromal cells, and the extracellular matrix (ECM). It can be divided into two broad phases: (1) Invasion of the ECM and (2) Vascular dissemination, homing, and colonization.
Robbins & Kumar Basic Pathology; Robbins, Cotran & Kumar Pathologic Basis of Disease
The Metastatic Cascade - Overview
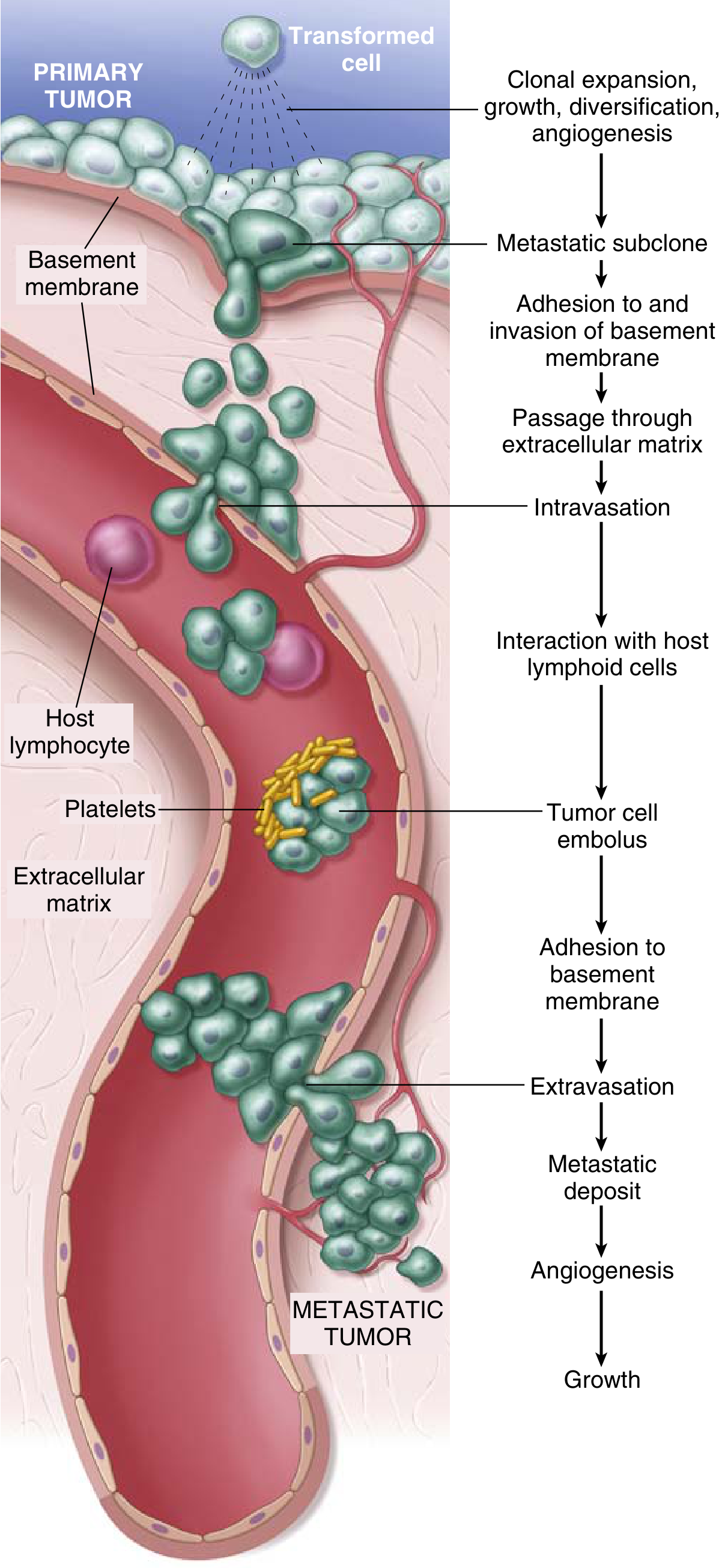
Fig. The metastatic cascade: sequential steps involved in hematogenous spread of a tumor - from clonal expansion at the primary tumor, through intravasation, tumor cell embolus formation, extravasation, and growth of the metastatic deposit.
The key steps are:
- Clonal expansion, growth, and angiogenesis at primary tumor
- Formation of a metastatic subclone
- Adhesion to and invasion of the basement membrane
- Passage through interstitial ECM
- Intravasation into blood/lymph vessels
- Interaction with host lymphoid cells and platelets (tumor cell embolus)
- Adhesion to vascular basement membrane at the distant site
- Extravasation
- Metastatic deposit formation, angiogenesis, and growth
PHASE 1: Invasion of the Extracellular Matrix
Human tissues are organized into compartments separated by two types of ECM: basement membranes and interstitial connective tissue, both composed of collagens, glycoproteins, and proteoglycans. ECM invasion occurs in four sequential steps:
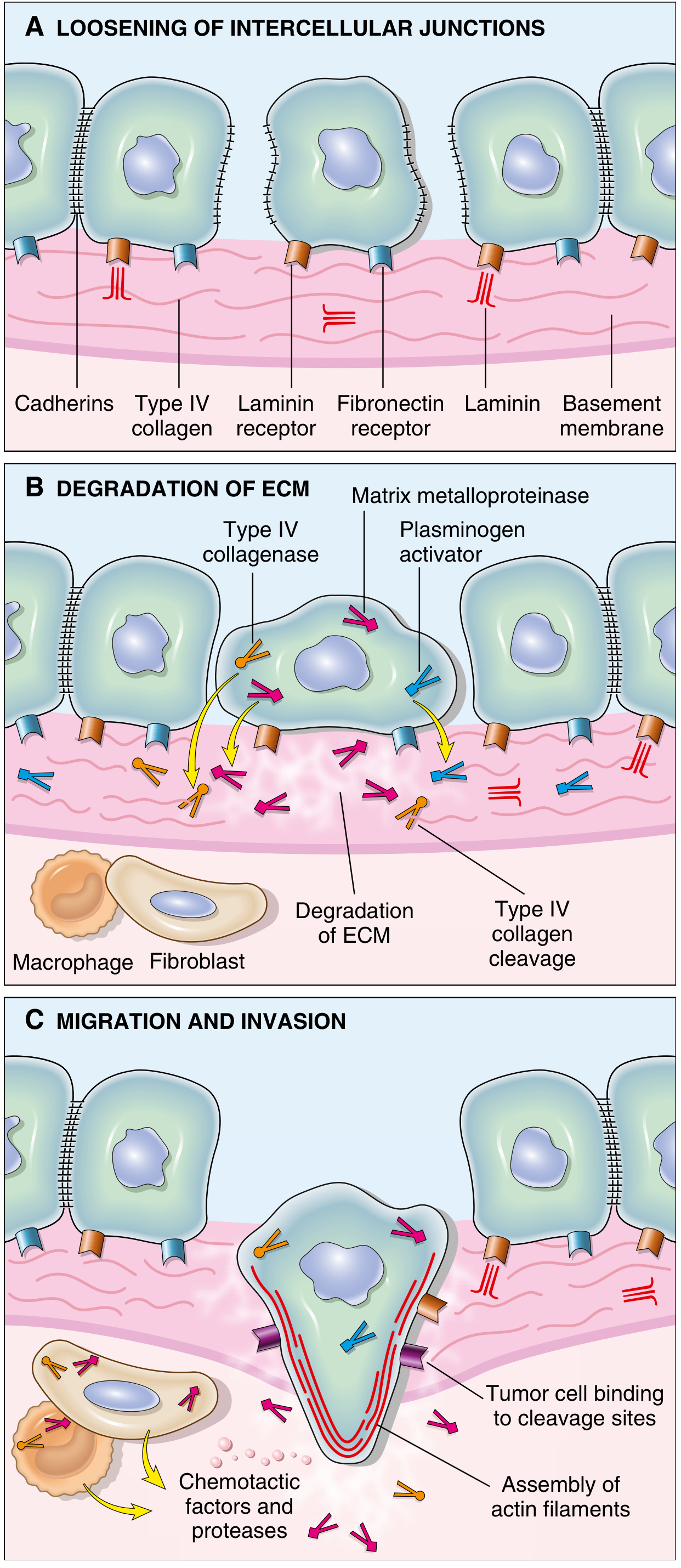
Fig. Sequence of events: (A) Loosening of intercellular junctions via cadherin loss; (B) Proteolytic degradation of ECM by MMPs, Type IV collagenase, and plasminogen activator; (C) Migration along cleavage sites driven by chemotactic factors and assembly of actin filaments.
Step 1 - Loosening of Cell-Cell Contacts
Normal epithelial cells are tightly held together by E-cadherins - transmembrane glycoproteins that mediate homotypic adhesion. Their cytoplasmic tails bind to beta-catenin, which in addition to its adhesive function also transmits anti-growth signals.
In tumors, E-cadherin function is lost by multiple mechanisms:
- Mutational inactivation of E-cadherin genes (e.g., certain adenocarcinomas of stomach and breast)
- Silencing via Epithelial-Mesenchymal Transition (EMT) - controlled by transcription factors SNAIL and TWIST
- Activation of beta-catenin genes, which disrupt cadherin-mediated cohesion
EMT is a key program implicated in the metastasis of carcinomas (particularly breast and prostate). It is defined not only by downregulation of epithelial markers (E-cadherin) but by upregulation of mesenchymal markers (vimentin, smooth muscle actin), together favoring a pro-migratory phenotype.
Step 2 - Degradation of the Basement Membrane and Interstitial ECM
Tumor cells degrade the structural barriers by secreting proteolytic enzymes directly or by inducing stromal cells (fibroblasts, inflammatory cells) to do so. Key enzymes include:
| Enzyme | Role |
|---|---|
| Matrix metalloproteinases (MMPs) | Remodel basement membrane and interstitial CT; MMP9 (gelatinase) cleaves type IV collagen |
| Cathepsin D | Lysosomal cysteine protease with ECM-degrading activity |
| Urokinase plasminogen activator (uPA) | Converts plasminogen to plasmin, which activates MMPs |
| Type IV collagenase | Directly cleaves the major component of epithelial basement membranes |
MMPs do far more than just cut a path - they also:
- Release VEGF from ECM-sequestered pools (promoting tumor angiogenesis)
- Generate chemotactic fragments from ECM proteins (attracting more tumor cells)
- Generate angiogenic cleavage products from collagen and proteoglycans
- Release growth factors (e.g., FGF, TGF-beta) embedded in the ECM
Importantly, concentrations of metalloproteinase inhibitors (TIMPs) are reduced in many cancers, further tilting the balance toward ECM destruction. Benign tumors show little MMP9 activity; their malignant counterparts overexpress it.
Cancer-associated fibroblasts (CAFs): Under the influence of invading cancer cells, stromal fibroblasts are reprogrammed (becoming CAFs) and alter expression of ECM molecules, proteases, protease inhibitors, and growth factors to support further invasion.
Step 3 - Attachment to Remodeled ECM Components
After degradation, new ECM binding sites are exposed. Tumor cells show complex changes in integrin expression - shifting from those that maintain normal tissue architecture to those that promote migration through the ECM. This step allows tumor cells to anchor and then propel themselves through the degraded matrix.
Laminin receptors and fibronectin receptors on tumor cell surfaces mediate attachment to the exposed cleavage products, facilitating directional movement.
Step 4 - Migration and Invasion
Locomotion is the final step, propelling tumor cells through degraded basement membranes and zones of matrix proteolysis. Migration involves:
- Attachment at the leading edge to ECM
- Detachment at the trailing edge
- Actin cytoskeleton contraction to ratchet the cell forward
Migration is stimulated and directed by:
- Tumor cell-derived autocrine factors: chemokines and growth factors (e.g., IGF-I, IGF-II)
- Cleavage products of matrix components: collagen and laminin fragments act as chemoattractants
- Stromal cell-derived paracrine factors: hepatocyte growth factor/scatter factor (HGF/SF) binds receptor tyrosine kinase MET on tumor cells and directly stimulates motility
This phase culminates in penetration through the endothelial basement membrane and transmigration into the vascular space - intravasation.
PHASE 2: Vascular Dissemination, Homing, and Colonization
Once in the bloodstream, tumor cells face hostile conditions: mechanical shear stress, immune attack (NK cells, CTLs), and anoikis (apoptosis triggered by loss of ECM contact).
Survival in Circulation
Despite these challenges, viable circulating tumor cells (CTCs) are not rare in patients with solid tumors. Several mechanisms enhance survival:
- Multicellular aggregation: Circulating cells that establish metastases are far more likely to migrate as multicellular aggregates than as single cells. Homotypic (tumor-tumor) and heterotypic (tumor-platelet) interactions both promote clumping.
- Platelet coating: Platelets surround tumor emboli (visible in the cascade diagram), enhancing survival in circulation by physically protecting cells from immune attack and shear stress.
- Fibrin deposition: Tumor cells expressing anionic substances (e.g., polyphosphate) activate Factor XII (contact factor), triggering coagulation and further stabilizing emboli in capillary beds.
- Stem cell-like subpopulations within emboli contribute relentless growth potential and phenotypic "plasticity" needed to adapt to foreign microenvironments.
Organ Tropism: Where Do Metastases Land?
The site of metastasis is determined by three factors:
1. Anatomical/vascular drainage (simple anatomy):
- Colon carcinoma → liver (portal circulation)
- Lung tumors → brain, adrenals, bone (systemic circulation)
- Most tumors arrest in the first capillary bed encountered (lung and liver most commonly)
2. Tumor cell tropism for specific tissues (the "seed and soil" hypothesis of Paget, 1889):
- Some tumors show organ tropism that cannot be explained by anatomy alone
- Prostate cancer preferentially metastasizes to bone; breast cancer to bone, brain, liver, lung
- This is mediated by expression of adhesion molecules or chemokine receptors on tumor cells whose ligands are expressed by endothelial cells of specific organs (e.g., CXCR4 on breast cancer cells, whose ligand CXCL12/SDF-1 is highly expressed in bone marrow)
3. Escape from tumor dormancy:
- Many micrometastases may be held in dormancy by immune surveillance or lack of angiogenesis
- Factors that break dormancy (e.g., loss of immune control, acquisition of angiogenic ability) allow outgrowth into clinically detectable deposits
Extravasation and Colonization at the Distant Site
To establish a secondary growth, circulating tumor cells must:
- Adhere to the vascular endothelium at the distant site (mediated by surface receptors)
- Extravasate - repeating the invasion process in reverse (breach the vascular basement membrane, invade the surrounding stroma)
- Establish a metastatic deposit - initially a micrometastasis
- Recruit blood supply (angiogenesis via VEGF and other signals)
- Grow into a macroscopic metastatic tumor
The metastatic microenvironment (the "soil") must be suitable. Tumor cells that survive must adapt to a tissue environment entirely different from the primary site, requiring phenotypic plasticity and often a collaboration between subclones.
Summary Table: Key Molecular Players
| Step | Key Molecules/Events |
|---|---|
| Cell-cell loosening | E-cadherin loss; EMT (SNAIL, TWIST); beta-catenin dysregulation |
| ECM degradation | MMPs (esp. MMP9), cathepsin D, uPA/plasmin, Type IV collagenase |
| ECM attachment | Integrins; laminin receptors; fibronectin receptors |
| Migration | HGF/MET axis; IGF-I/II; actin cytoskeleton remodeling; chemokines |
| Intravasation | VEGF-mediated angiogenesis (leaky vessels); endothelial transmigration |
| Survival in blood | Platelet coating; fibrin deposition; multicellular emboli |
| Organ tropism | CXCR4/CXCL12; adhesion molecule expression; "seed and soil" |
| Colonization | Angiogenesis; immune evasion; escape from dormancy |
EMT - A Unifying Mechanism
Epithelial-Mesenchymal Transition deserves special emphasis as a master regulatory event that links invasion to distant metastasis. During EMT:
- Epithelial markers (E-cadherin, cytokeratins) are downregulated
- Mesenchymal markers (N-cadherin, vimentin, fibronectin, smooth muscle actin) are upregulated
- SNAIL and TWIST transcription factors drive this reprogramming
- The result is a cell with increased motility, resistance to anoikis, stem-like properties, and resistance to chemotherapy
At the distant site, the reverse process - Mesenchymal-Epithelial Transition (MET) - may re-establish an epithelial phenotype to allow the cells to proliferate and form a cohesive secondary mass.
Key takeaways:
- Metastasis is an inefficient process - millions of cells enter the bloodstream daily, yet few produce clinically significant metastases
- No single "metastasis gene" has been found; instead, metastatic potential requires multiple complementary genetic and epigenetic alterations
- The tumor microenvironment (cancer-associated fibroblasts, inflammatory cells, endothelial cells) is an active participant, not a passive bystander
- A 2026 systematic review (PMID 41231587) on vasculogenic mimicry highlights an additional mechanism by which aggressive tumors form vessel-like structures without true endothelial cells, further supporting metastatic spread
Sources: Robbins & Kumar Basic Pathology (10th ed.), pp. 244-245; Robbins, Cotran & Kumar Pathologic Basis of Disease (10th ed.), pp. 290-292
This is a shared conversation. Sign in to Orris to start your own chat.