Explain action of aldosterone
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive, authoritative content from Ganong's and Brenner & Rector's Kidney to give a thorough answer. Let me compose the complete response.
Action of Aldosterone
Aldosterone is the principal mineralocorticoid secreted by the zona glomerulosa of the adrenal cortex. It plays a central role in sodium, potassium, water, and acid-base homeostasis.
1. Source and Basic Properties
- Synthesized exclusively in the zona glomerulosa via the enzyme aldosterone synthase (CYP11B2), which performs sequential 11β-hydroxylation, 18-hydroxylation, and 18-methyl oxidation of deoxycorticosterone.
- Secreted at 50-200 µg/day; plasma levels ~0.006 µg/dL (much lower than cortisol).
- Half-life ~20 min; poorly protein-bound.
(Brenner and Rector's The Kidney, p. 490)
2. Primary Target: The Aldosterone-Sensitive Distal Nephron (ASDN)
Aldosterone acts on the principal cells (P cells) of the:
- Distal convoluted tubule (DCT)
- Connecting tubule (CNT)
- Cortical collecting duct (CCD)
These segments express the mineralocorticoid receptor (MR) and the protective enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2).
3. Genomic (Classical) Mechanism of Action
Aldosterone is a steroid hormone and acts primarily through a nuclear receptor mechanism:
- Aldosterone diffuses into the principal cell and binds to the cytoplasmic mineralocorticoid receptor (MR).
- The aldosterone-MR complex translocates to the nucleus.
- It binds to mineralocorticoid response elements (MREs) on DNA.
- This alters transcription of target mRNAs, leading to synthesis of aldosterone-induced proteins (AIPs).
The key AIPs and their effects:
| Protein | Effect |
|---|---|
| SGK1 (serum- and glucocorticoid-regulated kinase 1) | Phosphorylates Nedd4-2, preventing it from ubiquitinating and removing ENaC from the membrane → increased apical ENaC expression |
| ENaC subunits (α, β, γ) | Aldosterone increases transcription of all three ENaC subunits; α-ENaC induction is rate-limiting (relieves a bottleneck in ENaC assembly and trafficking) |
| Na⁺-K⁺-ATPase (α₁ and β₁ subunits) | Increased expression on the basolateral membrane drives the Na⁺ gradient |
| CHIF (channel-inducing factor) | Further stimulates ENaC trafficking to the apical membrane |
The SGK1 gene is an early response gene - its mRNA rises within 15 min and protein within 30 min of aldosterone stimulation. This is why the peak effect on Na⁺ transport takes 10-30 minutes to develop and continues to build over hours.
(Ganong's Review of Medical Physiology, p. 358; Brenner and Rector's The Kidney, p. 502)
4. Net Renal Effect
Na⁺ reabsorption ↑ → K⁺ and H⁺ secretion ↑ → ECF volume expansion
- Apical ENaC reabsorbs Na⁺ from the tubular lumen into the principal cell.
- Basolateral Na⁺-K⁺-ATPase pumps Na⁺ into the interstitium (3 Na⁺ out, 2 K⁺ in).
- The electronegativity created in the tubular lumen drives K⁺ secretion via ROMK channels and H⁺ secretion via H⁺-ATPase on intercalated cells.
- Water follows Na⁺ passively → ECF volume expansion.
- Aldosterone also induces SLC26A4 (apical Cl⁻/HCO₃⁻ exchanger) on intercalated cells, contributing to acid-base regulation.
The graph below from Ganong's shows the time-course of these effects after a single aldosterone dose:
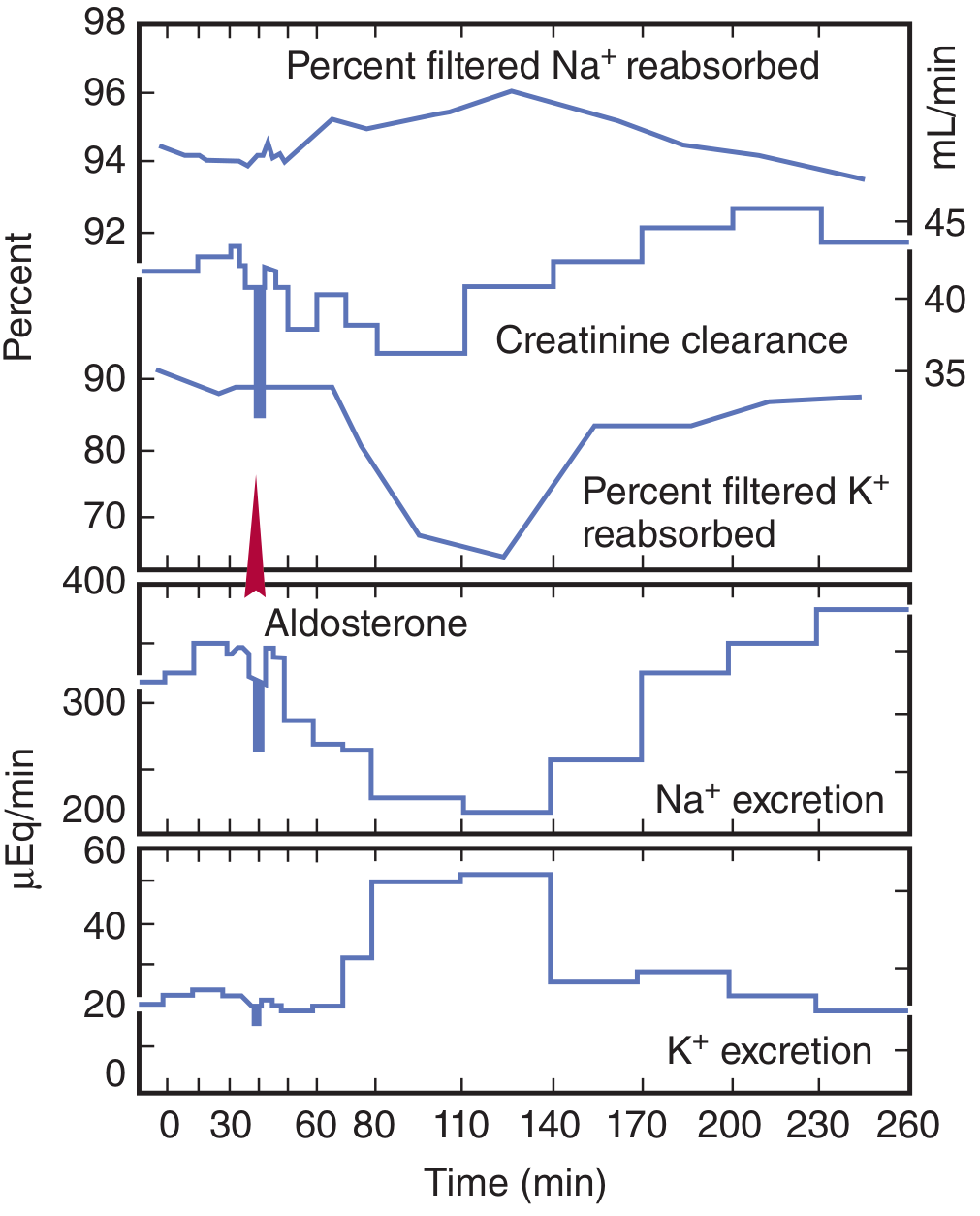
5. Non-Genomic (Rapid) Mechanism
In addition to its slow genomic action, aldosterone also binds to cell membrane receptors and rapidly (within minutes) increases the activity of membrane Na⁺-K⁺ exchangers through a non-genomic pathway. The second messenger is likely IP₃. However, this rapid effect is considered minor compared to the dominant genomic mechanism.
(Ganong's Review of Medical Physiology, p. 358)
6. Role of 11β-HSD2 (Specificity Guard)
The mineralocorticoid receptor (MR) has a higher affinity for cortisol than for aldosterone in vitro, and plasma cortisol is ~100-fold more abundant. To prevent cortisol from occupying MR and mimicking aldosterone:
- 11β-HSD2 in mineralocorticoid-sensitive tissues converts cortisol → cortisone (inactive at MR) and corticosterone → its 11-oxy derivative (also inactive).
- Aldosterone is left untouched by 11β-HSD2.
Clinical relevance: Inhibition of 11β-HSD2 (e.g., by licorice's glycyrrhetinic acid, or genetic deficiency) leads to Apparent Mineralocorticoid Excess (AME) - cortisol floods the MR causing hypertension and hypokalemia, with low plasma aldosterone and low renin (Clinical Box 19-3 - Ganong's).
7. Actions Beyond the Kidney
| Site | Effect |
|---|---|
| Sweat glands, salivary glands, colon | Increased Na⁺ reabsorption and K⁺ secretion (same ENaC-mediated mechanism) |
| Vascular wall | Vasoconstriction in response to volume depletion (expresses 11β-HSD2, so aldosterone-selective) |
| Nucleus tractus solitarius (brain) | Stimulates salt appetite; also expresses 11β-HSD2 |
| Taste buds | ENaC-mediated Na⁺ taste transduction |
(Brenner and Rector's The Kidney, p. 507)
8. Regulation of Aldosterone Secretion
Aldosterone secretion is primarily regulated by:
- Angiotensin II (RAAS activation in response to volume depletion/low BP) - most important
- Plasma K⁺ - direct stimulation of zona glomerulosa
- ACTH - minor role (short-term stimulation; important for the "aldosterone synthesis" pathway through StAR protein)
Summary Diagram of Mechanism
Volume depletion / ↑K⁺
↓
Angiotensin II / ACTH
↓
Adrenal Zona Glomerulosa
↓
ALDOSTERONE secreted
↓
Enters principal cell → binds cytoplasmic MR
↓
Aldosterone-MR complex → nucleus → binds MREs
↓
↑ Transcription of SGK1, ENaC subunits, Na⁺-K⁺-ATPase
↓
↑ Apical ENaC → Na⁺ reabsorption ↑
↑ Basolateral Na⁺-K⁺-ATPase → driving force ↑
↑ K⁺ secretion (ROMK) + H⁺ secretion (H⁺-ATPase)
↓
ECF volume expansion, ↓K⁺, metabolic alkalosis
Key clinical conditions related to aldosterone action:
- Primary hyperaldosteronism (Conn's syndrome): Hypertension, hypokalemia, metabolic alkalosis, low renin
- Hypoaldosteronism / Addison's disease: Na⁺ wasting, hyperkalemia, metabolic acidosis
- Pseudo-hypoaldosteronism: MR mutation - unresponsive to aldosterone
- Drugs: Spironolactone and eplerenone competitively antagonize the MR; amiloride/triamterene block ENaC directly
This is a shared conversation. Sign in to Orris to start your own chat.