Explain the patho physiology of MASLD
Reading File
Finding Sources
Finding Sources
Searching PubMed
MASLD "metabolic associated steatotic liver disease" pathophysiology
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
Reading File
I now have comprehensive content from Harrison's 22E on MASLD pathophysiology. I have everything I need to write a detailed, well-structured answer. Let me now compose the response.
Pathophysiology of MASLD (Metabolic Dysfunction-Associated Steatotic Liver Disease)
MASLD is the updated nomenclature (replacing NAFLD) that sits under the umbrella of steatotic liver disease (SLD). It is defined by hepatic steatosis plus one or more cardiometabolic risk factors, with minimal alcohol use (<20 g/d women; <30 g/d men). Its inflammatory subtype is MASH (replacing NASH), and the spectrum progresses from simple steatosis (MASL) through MASH to fibrosis and cirrhosis.
1. Core Concept: Energy Imbalance as the Upstream Driver
The foundational mechanism is an imbalance between energy intake and expenditure. Excessive caloric intake combined with reduced physical activity overwhelms the capacity of peripheral adipose tissue to store surplus energy. This leads to:
- Visceral adiposity
- Insulin resistance
- Systemic metabolic dysfunction
Harrison's Principles of Internal Medicine 22E, p. 2744
2. Hepatic Steatosis - The First Step
Triglyceride accumulates in hepatocytes (steatosis) through four main mechanisms:
| Mechanism | Details |
|---|---|
| Increased dietary FA delivery | High-fat, high-sugar diets increase free fatty acid (FFA) flux to the liver |
| Increased adipose lipolysis | Insulin resistance in adipocytes releases FFA into the portal circulation |
| De novo lipogenesis (DNL) | Dietary fructose and hyperinsulinemia upregulate DNL in hepatocytes |
| Impaired FA export/oxidation | Reduced VLDL secretion and impaired mitochondrial beta-oxidation trap fatty acids |
Triglyceride accumulation per se is a relatively safe storage form - steatosis alone does not damage the liver. However, it is a biomarker of underlying metabolic dysfunction that can progress further.
Genetic modifiers play a significant role:
- PNPLA3 I148M polymorphism - impairs lipolysis of lipid droplets; greatest known genetic risk factor
- TM6SF2 - influences cholesterol metabolism
- MBOAT7 - influences phospholipid metabolism
- HSD17B13 loss-of-function variants - protective against MASH, fibrosis progression, and liver cancer
Harrison's Principles of Internal Medicine 22E, p. 2744-2745
3. Transition to MASH (Steatohepatitis) - Lipotoxicity
The critical step distinguishing MASH from simple steatosis is lipotoxicity - lipid-associated cell injury.
How lipotoxicity develops:
- Dysregulated fatty acid processing produces toxic lipid intermediates (ceramides, diacylglycerols, lysophosphatidylcholine, reactive oxygen species)
- These intermediates activate endoplasmic reticulum (ER) stress, oxidative stress, and mitochondrial dysfunction
- Hepatocyte "resilience" (adaptive metabolic capacity) determines whether this resolves or progresses
Key mediators and pathways:
| Mediator | Role |
|---|---|
| Oxidative stress / ROS | Mitochondrial dysfunction generates free radicals that damage hepatocyte DNA, membranes, and proteins |
| ER stress (UPR activation) | Overwhelmed protein folding triggers inflammatory cascades (NF-κB, JNK activation) |
| Inflammatory cytokines | TNF-α, IL-6, IL-1β from activated Kupffer cells and adipose tissue promote hepatocyte injury |
| Hepatokines | Injured hepatocytes secrete altered signals (FGF21, fetuin-A) that alter systemic metabolism |
| Adipokines | Adiponectin (protective - decreased in obesity), leptin (pro-inflammatory - increased), resistin promote inflammation |
| Uric acid | Promotes oxidative stress and inflammasome (NLRP3) activation |
4. Role of Insulin Resistance
Insulin resistance is the central metabolic driver and operates at multiple levels:
- Adipocytes: Unrestrained lipolysis floods the portal circulation with FFAs
- Hepatocytes: Paradoxical insulin resistance - glucose homeostasis is impaired while insulin continues to drive DNL (selective insulin resistance - the lipogenic arm remains active)
- Pancreatic beta cells: Compensatory hyperinsulinemia further promotes fat synthesis and storage
Harrison's Principles of Internal Medicine 22E, p. 2745
5. Gut-Liver Axis
Obesity-associated changes in the gut microbiome contribute through two mechanisms:
- Enhanced energy harvest - altered microbiota extract more calories from dietary fiber
- Increased intestinal permeability - dysbiosis disrupts tight junctions, allowing bacterial products (LPS/endotoxin) to enter the portal circulation via the gut-liver axis
Hepatic toll-like receptors (TLR4) recognize LPS, triggering Kupffer cell activation and pro-inflammatory cytokine release (TNF-α, IL-1β), amplifying hepatic inflammation and steatohepatitis.
Harrison's Principles of Internal Medicine 22E, p. 2744; Benedé-Ubieto et al., Gut Microbes 2024
6. Fibrosis - The Determinant of Prognosis
Once lipotoxic stress is sustained, futile repair replaces healthy regeneration:
- Dying hepatocytes release DAMPs (danger-associated molecular patterns) that activate hepatic stellate cells (HSCs)
- Activated HSCs transdifferentiate into myofibroblasts and deposit collagen (fibrosis)
- Progressive fibrosis: pericellular "chicken-wire" fibrosis around zone 3 hepatocytes → bridging fibrosis (F3) → cirrhosis (F4)
- Fibrosis stage is the primary predictor of liver-related morbidity and mortality in MASLD
Fibrosis progression rates:
- Simple steatosis: ~1 stage per 14 years
- MASH: ~1 stage per 7 years
- Patients with MASH + fibrosis ≥F2 = "at-risk MASH" requiring intensive monitoring
7. Risk Factors That Amplify the Pathophysiology
| Risk Factor | Mechanism |
|---|---|
| Type 2 Diabetes | Strongest risk factor for MASLD acquisition, fibrosis progression, and HCC. Bidirectional - MASLD also increases T2DM risk 2-5x |
| Visceral/Android obesity | Increased visceral fat releases pro-inflammatory adipokines and FFAs |
| Dyslipidemia | Plasma lipid subfractions more atherogenic in MASLD (2x prevalence vs. non-MASLD) |
| Hypertension | Component of metabolic syndrome, amplifies vascular injury |
| High fructose/saturated fat diet | Upregulates DNL, promotes steatosis |
| Sedentary lifestyle | Reduces beta-oxidation in muscle and liver |
| Lean MASLD | Common in Asians - driven by increased visceral adiposity and insulin resistance despite normal BMI |
8. Histopathologic Spectrum
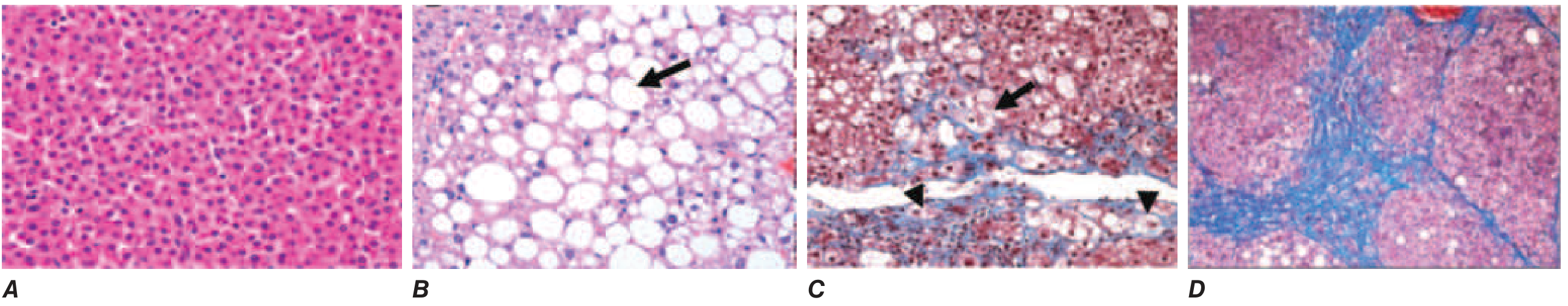
FIGURE 354-1 (Harrison's 22E): A. Healthy liver. B. Simple steatosis (fatty hepatocyte, arrow). C. MASH with ballooned hepatocyte (arrow) and pericellular fibrosis (arrowheads). D. Cirrhosis with bridging fibrosis.
Key histologic features of MASH:
- Macrovesicular steatosis (>5% hepatocytes)
- Hepatocyte ballooning (marker of lipotoxic injury; loss of keratin 8/18)
- Lobular inflammation (neutrophilic infiltrates)
- Pericellular fibrosis around zone 3 (centrilobular) hepatocytes - the "chicken-wire" pattern
- Note: Pediatric MASLD differs - shows more diffuse steatosis and portal (rather than central) fibrosis
9. Progression to HCC
Hepatocellular carcinoma (HCC) can occur both in the setting of cirrhosis (most common) and in non-cirrhotic MASLD. The mechanisms include:
- Sustained oxidative DNA damage
- Malignant transformation of regenerating hepatocytes under lipotoxic stress
- MASH-driven fibrosis creates a permissive oncogenic microenvironment
- Annual HCC incidence in MASLD-cirrhosis: 1-2% per year
Harrison's Principles of Internal Medicine 22E, Ch. 354
Summary Diagram of Pathogenesis
Energy excess + Sedentary lifestyle
↓
Visceral adiposity + Insulin Resistance
↓
↑ FFA flux → Hepatocyte + ↑ DNL + ↓ FA oxidation
↓
Triglyceride accumulation = STEATOSIS (MASL)
↓
Dysregulated FA processing → Toxic lipid intermediates
↓
LIPOTOXICITY
↓
Oxidative stress + ER stress + Inflammation + Gut dysbiosis (LPS/TLR4)
↓
Hepatocyte ballooning + Lobular inflammation = MASH
↓
Kupffer cell activation → Stellate cell activation → FIBROSIS
↓
Bridging fibrosis → CIRRHOSIS → HCC
Sources: Harrison's Principles of Internal Medicine 22E (2025), Chapter 354 (Manal F. Abdelmalek, Anna Mae Diehl) | Benedé-Ubieto et al., Gut Microbes 2024, PMID 38512763
Explain the patho physiology of MASLD
Reading File
Finding Sources
Searching PubMed
MASLD pathophysiology mechanisms
Reading File
Finding Sources
Reading File
Reading File
Loading Image
I now have all the key content needed. Let me compose the full, detailed response.
Pathophysiology of MASLD
MASLD (Metabolic Dysfunction-Associated Steatotic Liver Disease) replaced the older term NAFLD in 2023. It sits under the umbrella of steatotic liver disease (SLD) and is defined by hepatic steatosis + at least one cardiometabolic risk factor + minimal alcohol use (≤20 g/d women; ≤30 g/d men). Its inflammatory subtype is MASH (formerly NASH). The disease spans a spectrum from simple steatosis through steatohepatitis, fibrosis, cirrhosis, and HCC.
1. Upstream Driver: Energy Imbalance
The entire cascade is initiated by a mismatch between energy intake and expenditure. Excessive caloric intake + reduced physical activity overwhelms the capacity of peripheral adipose depots to store surplus energy, resulting in:
- Visceral adiposity
- Insulin resistance
- Systemic metabolic dysfunction
These three form the engine that drives everything downstream.
Harrison's Principles of Internal Medicine 22E (2025), Ch. 354
2. Hepatic Steatosis (MASL) - Step 1
Triglycerides accumulate inside hepatocytes through four converging mechanisms:
| Mechanism | Detail |
|---|---|
| ↑ Dietary fatty acid delivery | High-fat, high-sugar diets flood the portal circulation with free fatty acids (FFAs) |
| ↑ Adipose lipolysis | Insulin resistance in adipocytes removes the inhibitory brake on lipolysis, releasing FFAs into circulation |
| ↑ De novo lipogenesis (DNL) | Dietary fructose and hyperinsulinemia upregulate DNL within hepatocytes themselves |
| ↓ FA export / oxidation | Impaired VLDL secretion and reduced mitochondrial beta-oxidation trap fatty acids inside hepatocytes |
Triglyceride accumulation per se is a relatively safe storage form - it does not directly injure the liver. However, steatosis is a biomarker of underlying metabolic dysfunction that, left unchecked, sets the stage for lipotoxicity.
3. Transition to MASH (Steatohepatitis) - The Key Step: Lipotoxicity
Lipotoxicity (lipid-associated cell injury) is what distinguishes MASH from simple steatosis. It arises from dysregulated processing of fatty acids and lipid intermediates stored within lipid droplets, generating toxic by-products.
How lipotoxic stress develops:
Excess FFA → Dysregulated FA processing
→ Toxic lipid intermediates (ceramides, diacylglycerol,
lysophosphatidylcholine, reactive oxygen species)
→ ER stress, mitochondrial dysfunction, oxidative stress
→ Hepatocyte injury (ballooning, apoptosis, necroptosis)
Key mediators of lipotoxicity and inflammation:
| Mediator | Source | Effect |
|---|---|---|
| Reactive oxygen species (ROS) | Mitochondrial dysfunction | Oxidative damage to DNA, membranes, proteins |
| ER stress / Unfolded Protein Response | Overwhelmed hepatocyte proteostasis | Activates NF-κB and JNK → inflammation |
| TNF-α, IL-6, IL-1β | Activated Kupffer cells, visceral adipose | Pro-inflammatory hepatocyte injury |
| Adipokines | Adipose tissue | ↓ Adiponectin (anti-inflammatory, protective); ↑ Leptin, resistin (pro-inflammatory) |
| Hepatokines | Injured hepatocytes | Altered FGF21, fetuin-A secretion disrupts systemic metabolic crosstalk |
| Uric acid | Hepatocyte purine metabolism | Activates NLRP3 inflammasome; drives oxidative stress |
Hepatocyte resilience - the capacity to adapt metabolism to prevent/withstand/repair lipotoxic damage - is the key determinant of whether an individual develops MASH or recovers. Multiple genetic, epigenetic, and environmental factors modulate this resilience.
4. The Central Role of Insulin Resistance
Insulin resistance operates at multiple levels simultaneously, creating a self-amplifying loop:
- Adipocytes: Insulin fails to suppress lipolysis → unrestrained FFA release into portal blood
- Hepatocytes (selective/paradoxical IR): Glucose production is not suppressed (IR), but DNL remains insulin-responsive and is upregulated by compensatory hyperinsulinemia - this "selective IR" drives fat synthesis while glucose homeostasis fails
- Pancreatic beta cells: Compensatory hyperinsulinemia further promotes hepatic fat uptake, triglyceride synthesis, and fat storage
The MASLD-T2DM relationship is bidirectional: MASLD worsens insulin sensitivity and increases T2DM risk 2-5x, while T2DM is the single strongest risk factor for fibrosis progression and HCC in MASLD.
5. The Gut-Liver Axis
Obesity-associated gut dysbiosis contributes through two mechanisms:
- Enhanced energy harvest - altered microbiota ferment dietary fiber more efficiently, extracting additional calories
- Increased intestinal permeability - dysbiosis disrupts epithelial tight junctions, allowing bacterial lipopolysaccharide (LPS/endotoxin) to translocate into the portal circulation
LPS activates hepatic Toll-like receptor 4 (TLR4) on Kupffer cells, triggering NF-κB-mediated release of TNF-α and IL-1β, amplifying hepatic inflammation and steatohepatitis.
6. Genetics - Risk Modifiers
Several genetic polymorphisms substantially alter individual susceptibility:
| Gene | Variant | Effect |
|---|---|---|
| PNPLA3 | I148M (rs738409) | Impairs lipolysis of hepatic lipid droplets; the strongest known genetic risk factor for MASLD/MASH/HCC |
| TM6SF2 | E167K | Reduces VLDL secretion; promotes hepatic fat retention and fibrosis |
| MBOAT7 | rs641738 | Alters phospholipid remodeling; increases MASH and fibrosis risk |
| HSD17B13 | Loss-of-function | Protective against MASH, fibrosis progression, and liver cancer |
7. Fibrosis - The Determinant of Prognosis
When lipotoxic injury is sustained, futile repair replaces normal hepatic regeneration:
- Dying hepatocytes release DAMPs (danger-associated molecular patterns)
- DAMPs activate hepatic stellate cells (HSCs), which transdifferentiate into myofibroblasts
- Activated HSCs deposit collagen in a characteristic pericellular "chicken-wire" pattern around zone 3 hepatocytes
- Progressive fibrosis stages: pericellular (F1) → bridging (F3) → cirrhosis (F4)
Fibrosis stage is the single strongest predictor of liver-related morbidity and mortality in MASLD.
Fibrosis progression rates (from paired biopsy meta-analyses):
- Simple steatosis alone: ~1 fibrosis stage per 14 years
- MASH: ~1 fibrosis stage per 7 years
- MASH + fibrosis ≥F2 = "at-risk MASH" - the threshold for intensified monitoring and treatment
8. Progression to HCC
HCC can arise in both cirrhotic and non-cirrhotic MASLD livers (unlike most other causes of cirrhosis). Key oncogenic drivers include:
- Chronic oxidative DNA damage from sustained lipotoxic stress
- NLRP3 inflammasome activation
- Malignant transformation during dysregulated hepatocyte regeneration
- Fibrosis creating a permissive pro-oncogenic microenvironment
- Annual HCC incidence in established MASLD cirrhosis: 1-2% per year
9. Histopathologic Spectrum
The four panels below illustrate the full histopathologic progression:
A. Normal liver | B. Simple steatosis (arrow = fatty hepatocyte) | C. MASH - ballooned hepatocyte (arrow) with blue pericellular fibrosis (arrowheads) | D. Cirrhosis with bridging blue-stained fibrosis encircling regenerative nodules
Key histologic features of MASH:
- Macrovesicular steatosis (>5% of hepatocytes)
- Hepatocyte ballooning - swollen, pale cells with loss of keratin 8/18 (marker of lipotoxic injury)
- Lobular inflammation - neutrophilic and mixed infiltrates
- Pericellular fibrosis in zone 3 (centrilobular) - the hallmark "chicken-wire" pattern
Note: Pediatric MASLD differs - shows more diffuse steatosis, portal rather than centrilobular fibrosis, and ballooned hepatocytes may be absent
Summary: Pathogenesis at a Glance
Energy excess + Physical inactivity
↓
Visceral adiposity + Insulin Resistance
↓
↑ FFA flux to liver + ↑ DNL + ↓ beta-oxidation + ↓ VLDL export
↓
Hepatocyte triglyceride accumulation → STEATOSIS (MASL)
↓
Dysregulated FA metabolism → toxic lipid intermediates
(+ Gut dysbiosis → LPS/TLR4 → Kupffer cell activation)
(+ Genetic risk: PNPLA3, TM6SF2)
↓
LIPOTOXICITY → ER stress + ROS + Inflammation
↓
Hepatocyte ballooning + Lobular inflammation = MASH
↓
DAMP release → Hepatic stellate cell activation
↓
Collagen deposition → FIBROSIS
↓
Bridging fibrosis → CIRRHOSIS → HCC / Decompensation
Sources: Harrison's Principles of Internal Medicine 22E (2025), Chapter 354 (Abdelmalek & Diehl) | Steinberg et al., J Hepatol 2025 - integrative metabolism in MASLD/MASH | Miller et al., Endocrinol Diabetes Metab 2025 - mechanisms and therapeutics
This is a shared conversation. Sign in to Orris to start your own chat.