What is Gullain barre syndrome
Reading File
Finding Sources
Searching PubMed
"Guillain-Barre syndrome"[MeSH Terms]
Finding Sources
Reading File
Reading File
Reading File
Searching Images
Guillain-Barre syndrome nerve demyelination pathology
I now have comprehensive information from multiple authoritative textbooks. Let me compile the full answer.
Guillain-Barré Syndrome (GBS)
Definition
Guillain-Barré syndrome is an acquired, immune-mediated polyneuropathy of the peripheral nervous system characterized by rapidly progressive weakness, areflexia, and variable sensory involvement. It is one of the most common life-threatening diseases of the peripheral nervous system. — Robbins & Kumar Basic Pathology
The syndrome was first described in 1916 by Guillain, Barré, and Strohl, who identified its cardinal features: motor weakness, areflexia, paresthesias with minor sensory loss, and elevated CSF protein without pleocytosis (albumino-cytological dissociation). — Bradley and Daroff's Neurology in Clinical Practice
Epidemiology
- Incidence: approximately 1–2 per 100,000 per year
- Can occur at any age; slight male predominance
- Mortality has decreased dramatically from ~33% (before positive-pressure ventilation) to ~1–5% with modern critical care
Pathophysiology
About two-thirds of cases are triggered by a preceding infection, typically 1–4 weeks before neurological symptoms begin. The infection provokes microbe-specific T cells and antibodies that cross-react with peripheral nerve antigens (molecular mimicry) — causing the immune attack on nerve sheaths.
Key triggers include:
- Campylobacter jejuni (most common bacterial trigger — its lipooligosaccharides mimic GM1 gangliosides on myelin)
- Epstein-Barr virus (EBV)
- Cytomegalovirus (CMV)
- HIV
- Zika virus
- SARS-CoV-2
The injury is most extensive in nerve roots and proximal nerve segments, associated with mononuclear cell infiltrates rich in macrophages. Both T-cell–mediated and antibody-mediated mechanisms are involved. — Robbins & Kumar Basic Pathology
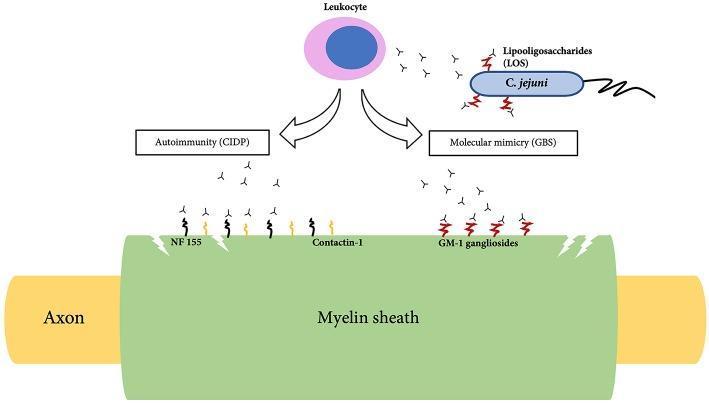
Pathophysiology of GBS: C. jejuni LOS triggers antibodies that cross-react with GM1 gangliosides on myelin (molecular mimicry), causing demyelination.
Subtypes
GBS is a heterogeneous syndrome — not all cases involve demyelination:
| Subtype | Description |
|---|---|
| AIDP (Acute Inflammatory Demyelinating Polyradiculoneuropathy) | Most common in Europe/North America; classic demyelinating form |
| AMAN (Acute Motor Axonal Neuropathy) | Pure motor axonal form; common in northern China, summer epidemics in children |
| AMSAN (Acute Motor-Sensory Axonal Neuropathy) | Axonal, involves motor + sensory fibers; severe with poor recovery |
| Miller-Fisher Syndrome (MFS) | Triad of ophthalmoplegia, ataxia, areflexia; anti-GQ1b antibodies; 6% of GBS in West, 18% in Taiwan |
| Acute Pandysautonomia | Combined sympathetic + parasympathetic failure without somatic involvement; anti-ganglionic ACh receptor antibodies |
| Pharyngeal-cervical-brachial variant | Bilateral cranial nerve palsies, especially bilateral facial palsy |
Clinical Features
Required for diagnosis:
- Progressive weakness of both legs and arms
- Areflexia or hyporeflexia
Supportive features:
- Progression over days to 4 weeks
- Relative symmetry
- Mild sensory symptoms
- Bifacial palsies
- Autonomic dysfunction (BP instability, arrhythmias, bladder dysfunction)
- Absence of fever at onset
- Recovery begins 2–4 weeks after progression stops
The typical course: ascending weakness starting in the legs → plateau (days to weeks) → gradual recovery. Symptoms usually reach their nadir within 2–4 weeks. — Robbins & Kumar
Diagnostic Criteria & Investigations
CSF (Lumbar Puncture):
- Classic finding: elevated protein with <10 cells/μL (albumino-cytological dissociation)
- May be normal in the first week in up to 50% of patients
- Moderate pleocytosis (>50 cells) is seen in HIV- or Lyme-associated GBS
Electrodiagnostic Studies (EDX/NCS/EMG):
- Abnormal in ~90% of established cases
- In AIDP: slowed conduction velocities, prolonged distal latencies, motor conduction blocks, absent or prolonged F-waves
- EDX may be normal early in the disease course
MRI (Spine with contrast):
- Shows enhancement and thickening of cauda equina nerve roots
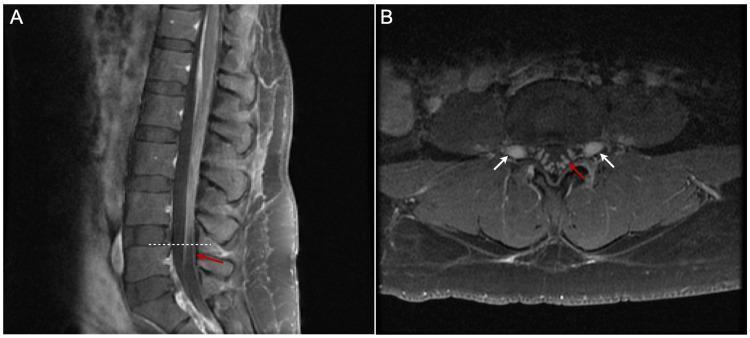
Post-contrast MRI: smooth thickening and linear enhancement of cauda equina nerve roots (red arrows) — classic for GBS/AIDP.
Antibodies (selected variants):
- Anti-GQ1b → Miller-Fisher Syndrome
- Anti-GM1, Anti-GD1a → AMAN
- Anti-ganglionic ACh receptor → Acute pandysautonomia
Treatment
- IVIG (Intravenous Immunoglobulin) — suppresses immune responses; standard first-line therapy
- Plasmapheresis (plasma exchange) — removes offending antibodies from circulation; equally effective to IVIG
- Supportive care — the most critical component:
- Mechanical ventilation if respiratory muscles are compromised (~25–30% of patients require intubation)
- Cardiac monitoring (autonomic instability)
- DVT prophylaxis, pain management, physical rehabilitation
- Corticosteroids are NOT effective in GBS (unlike CIDP)
Note: IVIG + plasmapheresis combined is not more effective than either alone.
Prognosis
- Most patients recover with time following the acute phase
- ~80% regain ability to walk independently within 6 months
- ~5–10% have severe residual disability
- ~1–5% mortality in modern ICU settings (mainly from respiratory failure, autonomic instability, or sepsis)
- Poor prognostic factors: older age, preceding C. jejuni infection, AMSAN subtype, need for mechanical ventilation, rapid disease progression
Differential Diagnosis
Conditions to exclude include: botulism, myasthenia gravis, transverse myelitis, critical illness neuropathy, tick paralysis, CIDP (chronic form), heavy metal poisoning, and Lyme disease.
Sources: Bradley and Daroff's Neurology in Clinical Practice; Robbins & Kumar Basic Pathology
This is a shared conversation. Sign in to Orris to start your own chat.