Congenital adrenal hyperplasia
Reading File
Finding Sources
Searching PubMed
"congenital adrenal hyperplasia"[MeSH Terms] AND management
Reading File
Reading File
Searching PubMed
"congenital adrenal hyperplasia"[MeSH Terms] AND treatment
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Reading File
Reading File
I now have comprehensive information. Let me compile the full answer.
Congenital Adrenal Hyperplasia (CAH)
Steroidogenesis Pathway
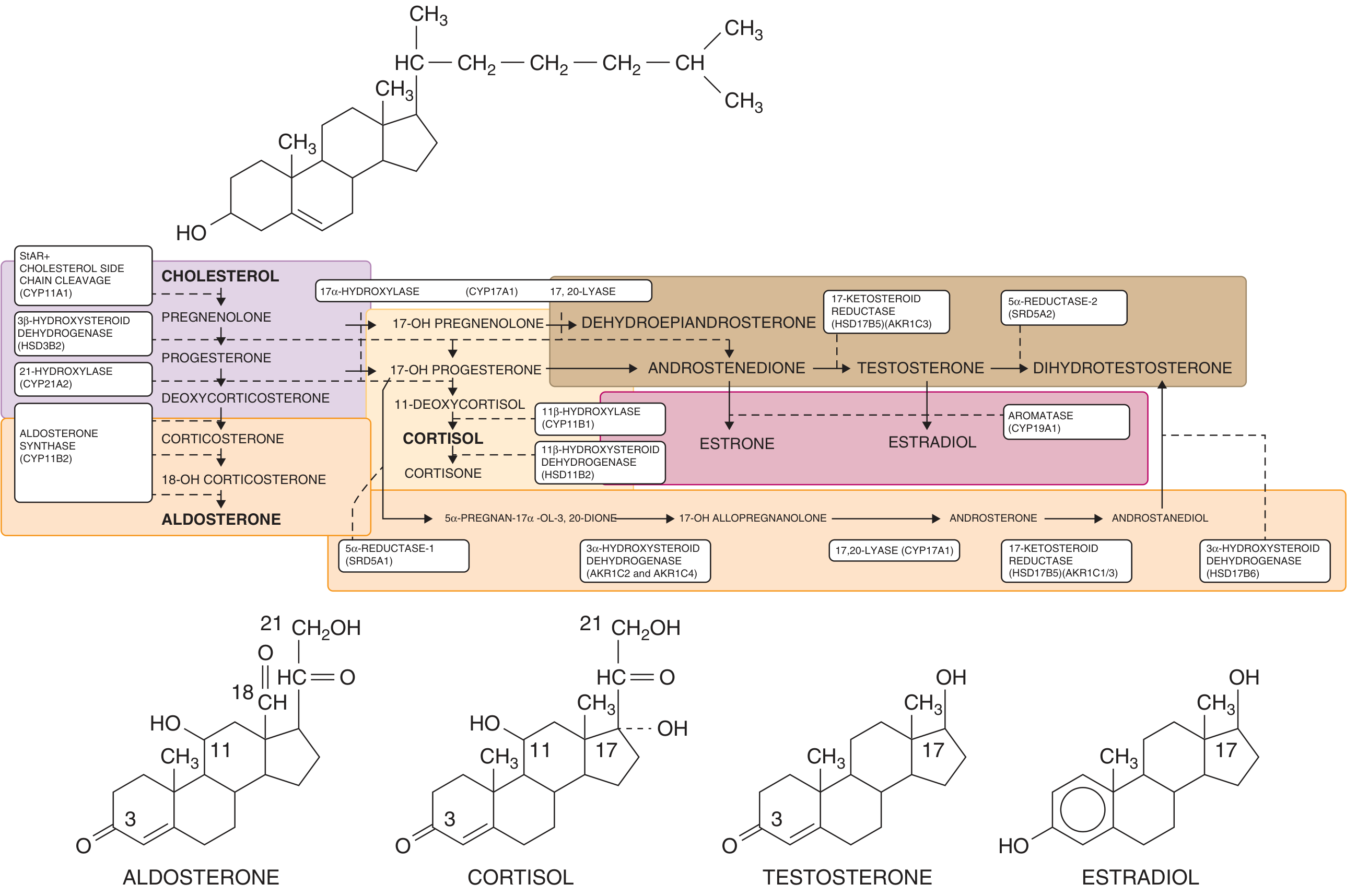
Adrenal and gonadal steroidogenesis. Enzyme blocks at key points divert precursors toward androgen synthesis. (Goldman-Cecil Medicine)
Definition and Pathophysiology
CAH is a group of autosomal recessive inborn errors of metabolism affecting cortisol biosynthesis. The fundamental defect is inadequate cortisol synthesis. This signals the hypothalamus and pituitary to increase CRH and ACTH, causing:
- Adrenocortical hyperplasia
- Accumulation of steroid precursors proximal to the enzymatic block
- Shunting of precursors toward androgen synthesis
Five enzymes can be defective, all involved in the cortisol biosynthetic pathway:
| Enzyme | Gene | Frequency | Key Features |
|---|---|---|---|
| 21-hydroxylase | CYP21A2 | ~95% of CAH | Virilization ± salt wasting |
| 11β-hydroxylase | CYP11B1 | ~5% | Virilization + hypertension |
| 17α-hydroxylase | CYP17A1 | Rare | Hypertension, sexual infantilism |
| 3β-HSD | HSD3B2 | Rare | Salt wasting, incomplete virilization |
| Cholesterol side-chain cleavage | CYP11A1 | Very rare | Lipoid adrenal hyperplasia |
21-Hydroxylase Deficiency (Most Common Form)
Genetics and Epidemiology
- Incidence: 1 in 5,000-15,000 in the US and Europe
- Highest incidence: 1 in 490 in Yupik Alaskan Eskimo population
- Gene locus: chromosome 6p21.3 within the HLA complex, transmitted autosomal recessively
- The CYP21A2 gene lies adjacent to an inactive pseudogene (CYP21PA1) that is 98% homologous - gene conversion during meiosis is a major mutational mechanism
- ~10 mutations account for 90-95% of alleles; over 200 different CYP21 mutations have been reported
Clinical Forms
1. Classic Salt-Wasting (75% of classic cases)
- Complete or near-complete 21-hydroxylase deficiency
- Aldosterone deficiency + cortisol deficiency + androgen excess
- Presents in neonates within 10-21 days of life: failure to thrive, vomiting, progressive weight loss, dehydration
- Life-threatening: hyperkalemia, hyponatremia, shock, adrenal crisis
- In males: often mistakenly diagnosed as pyloric stenosis or urosepsis
- In females: ambiguous genitalia at birth (always present since virilization begins at ~10 weeks gestation)
2. Classic Simple Virilizing (25% of classic cases)
- Partial 21-hydroxylase deficiency with residual aldosterone synthesis
- No salt wasting
- Females: virilization of external genitalia (clitoromegaly, labial fusion, urogenital sinus)
- Males: may appear normal at birth - diagnosis often delayed until signs of isosexual precocious puberty appear
3. Non-Classic (Attenuated) Form
- Mildest, most common form overall
- Often presents at puberty or adulthood
- Females: hirsutism, irregular menses, acne, short stature, oligomenorrhea (mimics PCOS)
- Males: may be asymptomatic or have acne/infertility
Prader Classification of Virilization (Females)
- Grades I-V reflecting progressive degrees of genital ambiguity, from clitoromegaly alone (I) to complete penile urethra (V)
11β-Hydroxylase Deficiency (~5% of CAH)
- Block at conversion of 11-deoxycortisol to cortisol and 11-deoxycorticosterone (DOC) to corticosterone
- DOC and 11-deoxycorticosterone accumulate - DOC has mineralocorticoid activity
- Features: virilization (like 21-OHD) plus hypertension (from DOC excess) and no salt wasting
- Hypokalemia may occur
Diagnosis
Newborn Screening
- Nationwide in all 50 US states and >40 countries
- Measures 17α-hydroxyprogesterone (17-OHP) on filter paper blood spot (elevated in 21-OHD)
- Has dramatically improved time to diagnosis and survival, especially in males with salt-wasting form
Laboratory Markers
| Test | Classic CAH | Non-Classic |
|---|---|---|
| Serum 17-OHP | Very high (>10,000 ng/dL) | Elevated (300-10,000 ng/dL after ACTH stim) |
| Plasma renin activity | Elevated (salt-wasting) | Normal/elevated |
| ACTH | Elevated | Elevated |
| Androgens (DHEA-S, androstenedione, testosterone) | Elevated | Mildly elevated |
| Electrolytes | Hyponatremia, hyperkalemia (salt-wasting) | Normal |
- ACTH stimulation test: 1 μg or 250 μg cosyntropin IV; 17-OHP >10,000 ng/dL at 60 min = classic CAH; 1,000-10,000 = non-classic
- Prenatal diagnosis: elevated 17-OHP or 21-deoxycortisol in amniotic fluid; genetic testing via CVS or amniocentesis
Treatment
Glucocorticoid Replacement
- Hydrocortisone: 10-20 mg/m²/day in 2-3 divided doses (first-line in children)
- Goal: suppress 17-OHP to 300-900 ng/dL (morning levels)
- Monitor: growth velocity, bone age, hormone levels - both overreplacement and underreplacement cause premature epiphyseal closure and short stature
- Adults: prednisolone or dexamethasone can be used (longer half-life, easier compliance), but dexamethasone carries higher risk of Cushingoid effects
Mineralocorticoid Replacement
- Fludrocortisone (Florinef): 0.05-0.2 mg/day orally
- Required in all patients with 21-OHD (even simple virilizers have subclinical aldosterone deficiency)
- Goal: suppress plasma renin activity to <5 ng/mL/hr
- Dietary salt supplementation in infants
Adrenal Crisis Management (Acute)
- Aggressive IV fluids (normal saline) for volume support
- Hydrocortisone IV: 1.5-2.0 mg/kg bolus, then 25-250 mg/day in divided doses
- Glucose supplementation
- Electrolyte correction
Stress Dosing ("Sick Day Rules")
- Double or triple the daily glucocorticoid dose during fever, illness, or surgery
- Parenteral hydrocortisone if unable to take oral medications
Surgical Management
- Females with ambiguous genitalia may need clitoral recession and vaginoplasty
- Timing is debated; the child must be of appropriate size for optimal outcome
Prenatal Treatment
- Dexamethasone given to the at-risk mother crosses the placenta and can reduce or prevent virilization of an affected female fetus
- Risks: maternal hypercortisolism, possible neurodevelopmental effects on infant
- Controversial - benefit only for affected females (~1/8 of all pregnancies at risk)
Complications and Long-term Issues
| Complication | Mechanism |
|---|---|
| Short stature | Premature epiphyseal closure from androgen excess or glucocorticoid overreplacement |
| Infertility | Anovulation from androgen excess (females); testicular adrenal rest tumors in males |
| Testicular adrenal rest tumors (TARTs) | Adrenal tissue in testes, can cause obstructive azoospermia |
| Osteoporosis | Glucocorticoid excess |
| Cardiovascular risk | Chronic glucocorticoid exposure, metabolic syndrome |
| Psychosocial issues | Gender identity concerns (especially 46,XX females with severe virilization), behavioral differences from prenatal androgen exposure |
| Adrenal insufficiency crisis | Inadequate stress dosing |
Key Points Summary
- CAH is the most common cause of ambiguous genitalia in 46,XX females
- 21-hydroxylase deficiency accounts for 95% of cases
- The classic triad in salt-wasting form: hyponatremia + hyperkalemia + elevated 17-OHP
- Salt-wasting form can be life-threatening in the first 2-3 weeks of life
- 11β-hydroxylase deficiency is the second most common and uniquely presents with hypertension (not salt wasting)
- Treatment is lifelong glucocorticoid + mineralocorticoid replacement
- All patients require stress dosing during illness or surgery
Sources: Goldman-Cecil Medicine, International Ed., Ch. 214; Campbell-Walsh-Wein Urology, 3-Vol. Set, Ch. 48; Berek & Novak's Gynecology; Miller's Anesthesia, 10th Ed.
This is a shared conversation. Sign in to Orris to start your own chat.