20yr male with severe vomiting due to motion sickness now not responding to call only response to painful stimuli
EMERGENCY: Altered Level of Consciousness After Severe Vomiting
Step 1 - Immediate Actions (First 5 Minutes)
| Priority | Action |
|---|---|
| Airway | Position in left lateral decubitus (recovery position) to prevent aspiration. Jaw thrust if needed. Consider immediate intubation (he cannot protect his airway) |
| Breathing | High-flow O2 via non-rebreather mask (15 L/min) |
| Circulation | IV access x2 large bore - draw bloods simultaneously |
| Disability | Formal GCS, pupil exam, blood glucose immediately |
| Exposure | Full skin/physical exam |
Step 2 - Differential Diagnosis
1. Severe Dehydration + Electrolyte Disturbance (Most Likely)
- Hyponatremia - loss of Na+ in vomitus + replacement with hypotonic fluids OR no intake. Acute hyponatremic encephalopathy causes cerebral edema and progresses to seizures, brainstem herniation, coma, and death. As Harrison's states: "Acute hyponatremic encephalopathy ensues when volume regulatory mechanisms are overwhelmed by a rapid decrease in tonicity, resulting in acute cerebral edema."
- Hypokalemia - severe K+ loss from vomiting
- Metabolic alkalosis - from H+ loss in gastric contents
- Hypoglycemia - no intake + repeated vomiting
2. Wernicke Encephalopathy (Thiamine Deficiency)
3. Medication Toxicity / Overdose
4. Aspiration + Hypoxia
5. Cerebral Event (must exclude)
6. Addisonian Crisis (Adrenal Insufficiency)
Step 3 - Immediate Investigations
| Investigation | Purpose |
|---|---|
| Bedside glucose | Rule out hypoglycemia first |
| ABG | O2 status, CO2, pH, electrolytes, lactate |
| Serum Na+, K+, Cl-, HCO3- | Critical - detect hyponatremia/hypokalemia |
| Serum osmolality | Hypo-osmolar vs hyperosmolar state |
| BUN/Creatinine | Renal function, dehydration |
| Blood glucose | Hypoglycemia / hyperglycemia |
| Full blood count | Infection |
| ECG | Hypokalemia-related arrhythmias |
| Urine ketones | Starvation ketosis |
| CT brain | If no obvious metabolic cause is found - must rule out bleed/herniation before LP |
| Thiamine level | If Wernicke suspected |
Step 4 - Treatment
Empirical Treatment (Do Not Delay for Results)
- Acute hyponatremia (symptomatic, comatose): 3% hypertonic saline - target raise Na+ by 1-2 mEq/L/hour for the first few hours (to stop cerebral edema) - but do not correct >8-10 mEq/L in first 24 hours (risk of osmotic demyelination)
- As Tintinalli's states: "When the patient presents with severe neurologic symptoms (vomiting, seizures, reduced consciousness), the most important guide for therapy is symptoms rather than the serum Na+"
Step 5 - Warning Signs That Change Management
| Finding | Action |
|---|---|
| Fixed/unequal pupils | Brain herniation - call neurosurgery, mannitol 0.5-1 g/kg IV |
| Fever + neck stiffness | Meningitis - LP and antibiotics STAT |
| "Thunderclap" headache history | SAH - urgent CT then LP |
| Seizures | Lorazepam 0.1 mg/kg IV + treat underlying cause |
| Severe hypotension | Sepsis or Addisonian crisis - consider hydrocortisone 100 mg IV |
| OphthalmopleGIA + ataxia | Wernicke - thiamine 500 mg IV TDS x 3 days |
Summary
- This patient needs emergency care NOW - he cannot protect his airway
- The most likely cause is acute electrolyte disturbance (hyponatremia/hypokalemia) from prolonged vomiting
- Wernicke encephalopathy must be treated empirically - give thiamine BEFORE glucose
- Do NOT give hypotonic IV fluids
- Check blood glucose at bedside within the first minute
- Consider intubation if GCS does not improve rapidly
- Harrison's Principles of Internal Medicine 22E (2025) - Hyponatremia, p. 2757-2764
- Goldman-Cecil Medicine - Wernicke Encephalopathy, p. 3586-3652
- Tintinalli's Emergency Medicine - Treatment of Electrolyte Disorders
Dose of paracetamol syrup 250/5 ml for 17kg child
Paracetamol (Acetaminophen) Dose for a 17 kg Child
Dose Calculation
| Parameter | Value |
|---|---|
| Standard dose | 10-15 mg/kg/dose |
| Child's weight | 17 kg |
| Dose range | 170 mg - 255 mg per dose |
| Practical dose (15 mg/kg) | 255 mg = ~5 mL |
| Conservative dose (10 mg/kg) | 170 mg = 3.4 mL |
Volume to Give
Dosing Schedule
| Frequency | Every 4-6 hours as needed |
| Maximum doses per day | 4-5 doses/day |
| Maximum daily dose | 75 mg/kg/day or 5 doses/day (whichever is less) |
| Max daily for 17 kg child | 17 × 75 = 1275 mg/day = 25.5 mL/day |
Practical Prescription
Paracetamol syrup 250 mg/5 mL Give 5 mL (250 mg) orally every 4-6 hours as needed for fever/pain. Do not exceed 5 doses in 24 hours.
- Do not give more frequently than every 4 hours
- Do not combine with other paracetamol-containing products (cold syrups, etc.)
- If fever persists >3 days or is >40°C, seek medical review
Bilateral cerbral hemisphere infarct with severe narrowing of basilar artery causes
bilateral cerebral hemisphere infarcts basilar artery stenosis causes etiology atherosclerosis vasculitis
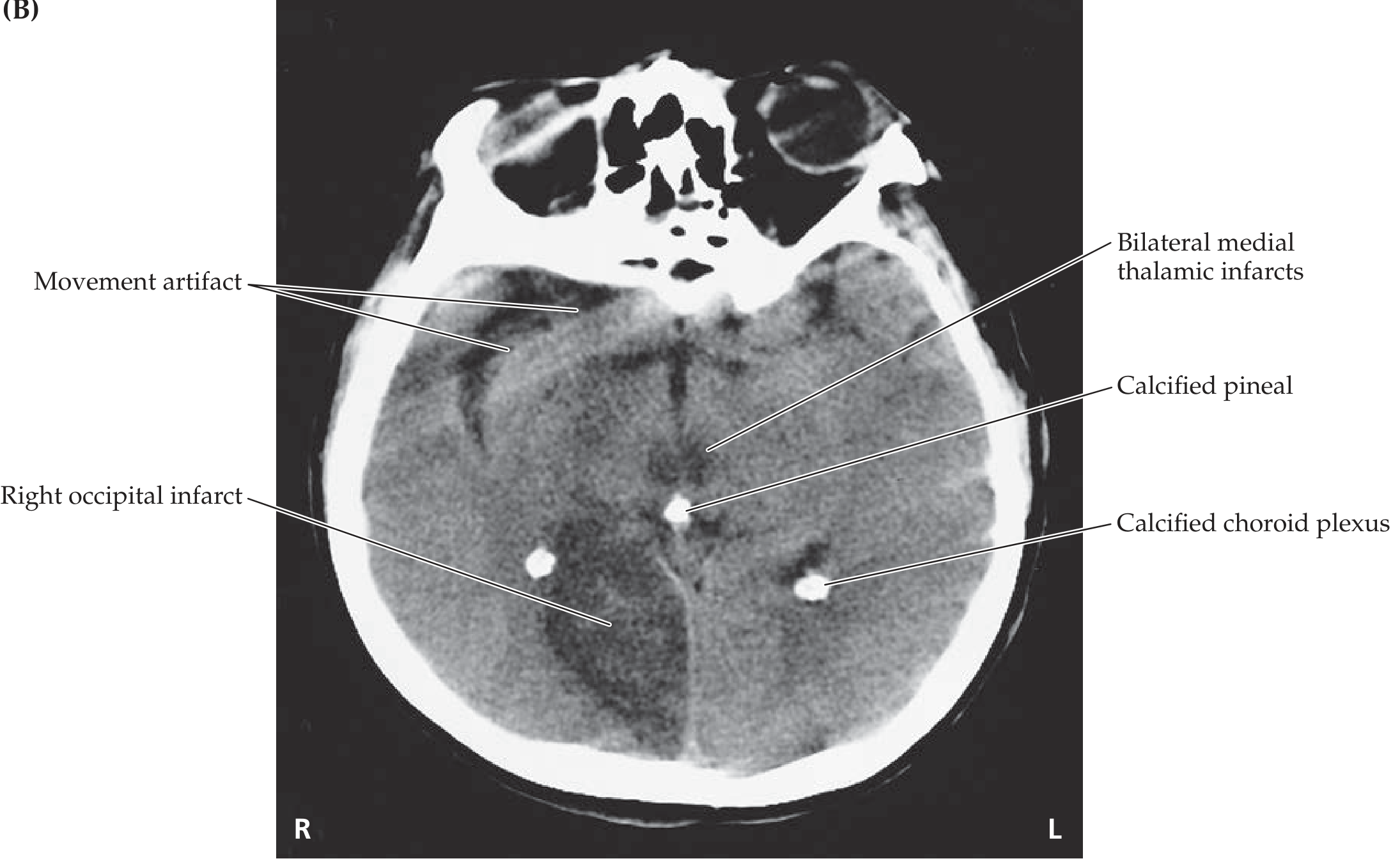
Bilateral Cerebral Hemisphere Infarcts + Severe Basilar Artery Narrowing - Causes
Understanding the Anatomy First
- Posterior cerebral arteries (PCAs) - supplying medial temporal and occipital lobes
- Thalamo-perforating arteries - bilateral thalamic supply
- Superior cerebellar arteries
- Pontine perforators
Causes - Classified by Mechanism
1. Intracranial Atherosclerosis (Most Common Overall)
- In-situ thrombosis on the plaque surface
- Artery-to-artery embolism - plaque ruptures and debris embolizes distally to bilateral PCAs
- Branch occlusion - plaque blocks origins of perforating arteries
2. Cardioembolism
| High-Risk Source | Notes |
|---|---|
| Atrial fibrillation | Most common cause of cardioembolism |
| Recent MI with LV thrombus | 85% of emboli within first 4 weeks |
| Dilated cardiomyopathy | Stasis and thrombus formation |
| Infective endocarditis | Septic emboli - multiple territory |
| Mitral stenosis (rheumatic) | Emboli in 9-14% of patients |
| Patent foramen ovale | Paradoxical embolism |
| Prosthetic heart valves | |
| Atrial myxoma |
3. "Top-of-Basilar" Syndrome
- Bilateral occipital lobe infarcts (cortical blindness)
- Bilateral thalamic infarcts (altered consciousness, memory loss)
- Midbrain infarcts (somnolence, diplopia, abnormal pupils)
- The basilar artery itself may appear narrowed from the embolus or underlying atherosclerosis
4. Vertebral Artery Dissection
5. Vasculitis - Inflammatory/Autoimmune
| Vasculitis | Key Features |
|---|---|
| Primary CNS vasculitis (PACNS) | Isolated to CNS; multi-territory strokes in young adults |
| Giant Cell Arteritis (GCA) | Age >50; jaw claudication, scalp tenderness; affects VA and basilar |
| Takayasu arteritis | Young women; affects aorta and branches |
| Behçet disease | Oral/genital ulcers; can cause basilar thrombosis |
| Neurosyphilis (meningovascular) | Progressive arteritis; affects basilar artery classically |
| Neuroborreliosis | Lyme disease; 8 of 11 cases in one series involved vertebrobasilar circulation |
| CNS lupus (SLE) | Immune-mediated vasculopathy |
6. Hypercoagulable States
- Antiphospholipid syndrome - recurrent strokes in young patients; anti-cardiolipin antibodies
- Polycythemia vera - hyperviscosity
- Essential thrombocythemia
- Protein C / Protein S / Antithrombin III deficiency
- Factor V Leiden mutation
- Nephrotic syndrome (hypercoagulable from loss of anticoagulant proteins)
- Malignancy (Trousseau syndrome) - mucin-secreting adenocarcinomas
7. Basilar Artery Dolichoectasia
8. Hemodynamic (Watershed) Infarction
9. Genetic / Metabolic Causes (Rare)
- MELAS (mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes) - stroke-like episodes NOT in vascular territory; bilateral; associated with seizures, lactic acidosis
- CADASIL (cerebral autosomal dominant arteriopathy) - young adults, leukoencephalopathy
- Fabry disease - alpha-galactosidase A deficiency; preferentially affects posterior circulation
Key Investigative Approach
| Investigation | What It Detects |
|---|---|
| MRI brain + DWI | Confirms infarct location and age |
| MRA/CTA brain and neck | Basilar artery stenosis, vertebral dissection |
| Vessel wall MRI | Differentiates atherosclerosis vs vasculitis (wall enhancement = vasculitis) |
| Echocardiogram (TTE/TEE) | Cardiac source of embolism |
| Holter / telemetry | Paroxysmal AF |
| Lipid panel, HbA1c, BP | Atherosclerotic risk factors |
| Hypercoagulable screen | APS antibodies, protein C/S, Factor V Leiden, etc. |
| ESR, CRP, ANCA, ANA | Vasculitis screen |
| Blood cultures / VDRL | Infective endocarditis, syphilis |
| Serum/CSF lactate | MELAS |
Summary
| Mechanism | Key Cause | Clue |
|---|---|---|
| Large vessel atherosclerosis | Hypertension, diabetes, smoking | Older patient, gradual onset |
| Cardioembolism | AF, MI, cardiomyopathy | Bilateral large infarcts, sudden onset |
| Top-of-basilar embolism | Embolic (any source) | Cortical blindness + thalamic signs |
| Vasculitis | GCA, PACNS, syphilis, Behçet | Young patient, progressive, CSF pleocytosis |
| Hypercoagulable | APS, polycythemia | Recurrent strokes, young patient |
| Dissection | VA dissection | Young, neck trauma/manipulation |
Can wernikes encephalopathy or severe dehydration due to vomiting cause this condition
Wernicke encephalopathy cerebral infarction basilar artery
dehydration vomiting cerebral venous thrombosis ischemic stroke
Wernicke encephalopathy basilar artery stenosis cerebral infarct mechanism vomiting
severe dehydration vomiting cerebral venous thrombosis bilateral infarct mechanism hyperviscosity
Can Wernicke Encephalopathy or Severe Dehydration from Vomiting Cause Bilateral Cerebral Infarcts + Basilar Artery Narrowing?
1. Wernicke Encephalopathy (WE)
Can it directly cause cerebral infarcts? - NO (not true arterial infarcts)
Can it mimic infarcts on imaging? - YES, powerfully
Thiamine is an essential cofactor for energy metabolism. Its deficiency causes selective failure of oxidative metabolism in high-demand brain regions - not arterial occlusion.
| Region | Why vulnerable |
|---|---|
| Medial thalami (bilateral) | High metabolic demand, thiamine-dependent enzymes |
| Mammillary bodies | Classically affected |
| Periaqueductal grey matter | Surrounds cerebral aqueduct, high activity |
| Superior and inferior colliculi | |
| Floor of fourth ventricle |
The critical imaging trap:
- WE causes bilateral medial thalamic signal changes on MRI that look exactly like bilateral thalamic infarcts
- The DWI restriction (bright on diffusion-weighted imaging) mimics acute ischemic infarct
- This is the same pattern as "top-of-basilar" syndrome (bilateral thalamic + occipital infarcts from basilar apex occlusion)
- It is very easy to misdiagnose WE as bilateral PCA territory infarction
Does WE cause basilar artery narrowing? - NO
- A coincidence (the stenosis is the actual cause of thalamic infarcts)
- Artefact on MRA from slow/turbulent flow rather than true stenosis
WE can be caused by vomiting - YES
- Hyperemesis gravidarum
- Malignant GI obstruction
- Prolonged vomiting from any cause (including our motion sickness patient)
2. Severe Dehydration from Vomiting
Can it cause bilateral cerebral infarcts? - YES, via several mechanisms
A. Cerebral Venous Sinus Thrombosis (CVST) - Most important mechanism
"Most often, this (venous sinus thrombosis) occurs during a hypercoagulable state related either to dehydration, infection, or childbirth."
- Bilateral thalamic venous infarcts (can look like bilateral deep infarcts)
- Altered consciousness progressing to coma
- Nausea and vomiting (both a cause and symptom)
B. Hemodynamic/Watershed Infarction
C. Hyponatremia-induced Cerebral Edema (not infarction, but mimics it)
Can dehydration narrow the basilar artery? - INDIRECTLY
- Hemoconcentration + hypotension in the setting of pre-existing atherosclerotic plaque can trigger in-situ thrombosis on a narrowed basilar artery, making it appear severely stenosed on imaging
- MRA performed during dehydration/hypotension may show apparent "narrowing" due to slow or reduced flow rather than true stenosis (flow-related artifact)
3. The Combined Picture - Putting It Together
Severe Vomiting
│
├── Thiamine depletion → WERNICKE ENCEPHALOPATHY
│ └── Bilateral thalamic metabolic necrosis
│ (mimics bilateral thalamic infarcts on MRI)
│
├── Dehydration → CVST
│ └── Bilateral thalamic venous infarcts
│ (especially with deep venous thrombosis)
│
├── Hypovolemia + pre-existing basilar stenosis
│ └── Hemodynamic infarcts in posterior territory
│
└── Hyponatremia
└── Cerebral edema → secondary ischemia
Clinical Bottom Line
| Condition | Causes true infarcts? | Mimics infarcts? | Affects basilar? | Key distinguishing feature |
|---|---|---|---|---|
| Wernicke encephalopathy | No (metabolic necrosis) | Yes - bilateral thalami, DWI+ | No | Responds to IV thiamine; mammillary body involvement; no vascular distribution |
| Dehydration (CVST) | Yes (venous infarcts) | - | Indirectly | Non-arterial distribution; hemorrhagic transformation; MR venogram confirms |
| Dehydration + pre-existing basilar stenosis | Yes (hemodynamic) | - | Triggers thrombosis | Posterior circulation territory; prior TIA symptoms |
| Hyponatremic encephalopathy | Secondary only | Yes (diffuse edema) | No | Symmetric; reverses with Na+ correction |
Most important clinical action:
Seizure and excessive vomiting with blood content in middle aged man after excessive alcohol consumption
alcohol withdrawal seizures hematemesis Mallory Weiss tear management emergency department
Seizure + Bloody Vomiting After Excessive Alcohol Consumption - Middle-Aged Man
- Seizure - almost certainly alcohol withdrawal
- Hematemesis (bloody vomiting) - most likely Mallory-Weiss tear, less likely variceal bleed
PART 1 - THE SEIZURE
Cause: Alcohol Withdrawal Syndrome (AWS)
Timeline of Alcohol Withdrawal
| Time After Last Drink | Syndrome | Features |
|---|---|---|
| 6-12 hours | Minor withdrawal | Anxiety, tremor, nausea, vomiting, sweating, tachycardia |
| 12-24 hours | Alcoholic hallucinosis | Visual/auditory/tactile hallucinations with intact orientation |
| 12-48 hours | Withdrawal SEIZURES | Generalized tonic-clonic; typically single or cluster |
| 48-72 hours | Delirium Tremens (DT) | Confusion + autonomic storm; most dangerous; mortality up to 5-15% untreated |
- Seizures occur in 5-10% of AWS patients
- Usually generalized tonic-clonic
- Two-thirds of patients who have one untreated seizure will have more
- 3% develop status epilepticus
- Most cluster at 6-48 hours after the last drink
Other Causes of Seizure to Consider in This Patient
| Cause | Key Clue |
|---|---|
| Hypoglycemia | Check glucose immediately - alcoholics skip meals |
| Hyponatremia | Excessive water/beer intake with poor nutrition |
| Wernicke encephalopathy | Thiamine deficiency from poor diet |
| Head trauma | Intracranial bleed from alcohol-related fall |
| Hepatic encephalopathy | Elevated ammonia; asterixis; jaundice; confusion |
| Meningitis | Fever + neck stiffness; alcoholics are immunocompromised |
| Drug co-ingestion | Stimulants, cocaine, benzodiazepine co-use |
PART 2 - BLOODY VOMITING (HEMATEMESIS)
Most Likely Cause: Mallory-Weiss Tear
"Longitudinal mucosal tears near the gastroesophageal junction, called Mallory-Weiss tears, are most most often associated with severe retching or vomiting secondary to acute alcohol intoxication."
Non-bloody vomiting/retching first → then hematemesis
- Accounts for 10-15% of upper GI bleeds
- Mortality ~5%
- Bleeding stops spontaneously in most cases
- Worsened significantly if patient has underlying cirrhosis/portal hypertension
Second Most Likely: Esophageal / Gastric Varices
- Accounts for the vast majority of GI bleeding in cirrhotics
- Massive, life-threatening hematemesis (often bright red, large volume)
- 50% of cirrhotics develop varices; 10-15% bleed per year
- Look for: spider angiomata, jaundice, ascites, caput medusae, palmar erythema, splenomegaly
Other Causes of Hematemesis
| Cause | Notes |
|---|---|
| Peptic ulcer disease (PUD) | Alcohol + NSAIDs damage gastric mucosa |
| Acute alcoholic gastritis | Diffuse mucosal erosions from alcohol toxicity |
| Boerhaave syndrome | Full-thickness esophageal rupture from severe retching - SURGICAL EMERGENCY; presents with chest pain + shock |
| Dieulafoy lesion | Submucosal artery erosion; rare but massive bleed |
| Esophagitis | Erosive; less severe bleeding |
EMERGENCY MANAGEMENT
Immediate Priorities (Simultaneous)
AIRWAY first - this patient has seizures + vomiting = extreme aspiration risk
Intubation if GCS low, ongoing seizures, or massive hematemesis
Step-by-Step
| Priority | Action |
|---|---|
| A - Airway | High aspiration risk (vomiting + seizures). RSI/intubate if not protecting airway |
| B - Breathing | O2 15 L/min non-rebreather |
| C - Circulation | 2x large-bore IV access; blood for FBC, coagulation, LFTs, U&E, glucose, blood group & crossmatch, ammonia, blood cultures |
| D - Dextrose | Bedside glucose STAT - treat hypoglycemia if present |
| Thiamine 200 mg IV | Give BEFORE glucose - prevents Wernicke encephalopathy |
| Seizure control | Lorazepam 0.1 mg/kg IV or diazepam 10 mg IV - benzodiazepines are first-line for alcohol withdrawal seizures |
| Hematemesis | IV PPI (pantoprazole 80 mg bolus then 8 mg/hr); if variceal suspected: octreotide/terlipressin + ceftriaxone 1g IV |
| Fluid resuscitation | Normal saline; blood transfusion if Hb <7 g/dL (or <8 if variceal) |
Why Benzodiazepines for Seizure?
Investigations
| Test | Looking for |
|---|---|
| Glucose (bedside) | Hypoglycemia |
| FBC | Anemia from bleeding; thrombocytopenia (cirrhosis/alcohol) |
| Coagulation (PT/INR) | Liver synthetic failure; coagulopathy worsens bleeding |
| LFTs, albumin | Liver disease severity |
| Ammonia | Hepatic encephalopathy |
| Serum Na+, K+, Mg2+ | Electrolyte disturbances (all common in alcoholics) |
| CT brain | If seizure atypical or focal, or head trauma suspected |
| Urgent endoscopy (EGD) | After resuscitation and airway secured - identifies bleeding source |
Endoscopy (EGD)
- Mallory-Weiss tear: treat with epinephrine injection, clips, or band ligation
- Varices: band ligation is first-line endoscopic treatment
- If varices cannot be controlled: Sengstaken-Blakemore tube tamponade as bridge to TIPS (transjugular intrahepatic portosystemic shunt)
Summary Diagram
Middle-aged man + alcohol + vomiting + seizure + hematemesis
│ │
ALCOHOL WITHDRAWAL MALLORY-WEISS TEAR
(12-48h after last drink) (forceful retching → GEJ tear)
│ │
Benzodiazepines Endoscopy + PPI
Thiamine first!
Check glucose
│
± VARICEAL BLEED
(if cirrhosis present)
Octreotide + antibiotics + banding
- Thiamine before glucose
- Benzodiazepines - not phenytoin - for withdrawal seizures
- Intubate early if airway compromised - vomiting + seizures = fatal aspiration
- Alcohol withdrawal seizures often recur within hours - observe minimum 6 hours
If a child comes with history breakge of glass while having water and parents are suspecting accidental ingestion of the small particles of glass
child accidental glass ingestion management radiology observation guidelines safe to pass
Child with Suspected Glass Particle Ingestion
First - Reassure the Parents (Appropriately)
Step 1 - Immediate Assessment
Ask These Questions First
| Question | Why It Matters |
|---|---|
| Is the child symptomatic RIGHT NOW? | Drooling, choking, stridor = esophageal/airway emergency |
| How much glass / what size particles? | Tiny powder fragments vs. large shard = very different risk |
| Was it witnessed or just suspected? | May need imaging to confirm ingestion at all |
| Where is the pain, if any? | Throat/chest = esophageal; abdomen = stomach/bowel |
| Any blood in mouth, saliva, or vomit? | Mucosal injury already present |
| Any difficulty breathing? | Airway involvement / aspiration |
Red Flag Symptoms - Require Immediate Action
- Stridor, drooling, inability to swallow
- Neck or chest pain
- Vomiting blood (hematemesis)
- Abdominal pain or rigidity
- Respiratory distress
- Coughing/choking that won't settle
Step 2 - Why Glass Is a Special Case
- A normal X-ray does NOT rule out glass ingestion
- Standard coin/foreign body protocols do not fully apply
- If symptomatic, CT is needed
"CT scanning is a very high-yield test for esophageal foreign body and has generally replaced the barium swallow to evaluate ingestion of non-radiopaque objects."
"Radio-lucent + symptomatic witnessed ingestion: Urgent endoscopic evaluation and removal. Asymptomatic: Consider CT, ultrasound, MRI or esophagram for further assessment."
- Plain X-ray did NOT clearly identify the glass
- CT scan was required to locate it (found in gastric antrum)
- Conservative management was successful with vigilant monitoring
Step 3 - Management Algorithm
Child with suspected glass ingestion
│
┌─────────┴──────────┐
SYMPTOMATIC ASYMPTOMATIC
│ │
Emergency assessment │
Airway/ABC first │
IV access │
Surgical consult │
Urgent endoscopy │
┌───────┴────────┐
Very small particles Larger/sharp fragment
(powder/dust-like) (visible shard suspected)
│ │
Observe; educate parents CT scan to locate
Reassurance appropriate + Surgical/GI consult
Watch stools + Likely endoscopy
If Asymptomatic with Tiny Glass Particles (Most Common Scenario)
- Often pass through GI tract without causing mucosal injury
- Drinking water naturally smooths micro-edges during transit
- Self-limited in most published cases
- Thorough oral cavity examination - check mouth, gums, tongue for cuts
- Neck and abdominal exam - tenderness, crepitus, distension
- Plain X-ray (neck/chest/abdomen) - glass may not show but rules out other findings (free air = perforation)
- If asymptomatic and tiny particles suspected - conservative observation is reasonable
- Educate parents on warning signs to return
If a Larger Shard Is Suspected or Child is Symptomatic
| Location | Action |
|---|---|
| Esophageal (drooling, dysphagia, chest pain) | Urgent endoscopic removal within 2 hours |
| Gastric (in stomach) | Endoscopic removal - sharp objects in stomach should generally be removed promptly regardless of size (Yamada's Gastroenterology) |
| Past pylorus, asymptomatic | Serial X-rays/CT to confirm progression; return if symptoms develop |
| Past pylorus, symptomatic | Surgical/enteroscopy consultation |
"Ingestion of sharp or elongated objects" is a standalone indication for urgent endoscopy
Step 4 - What to Tell the Parents
Safe discharge with strict return precautions if:
- Child is completely asymptomatic
- No oral/pharyngeal cuts visible
- No abdominal tenderness
- Particles were described as tiny/powder-like
- X-ray shows no free air or obvious object
Return immediately if child develops:
- Drooling or difficulty swallowing
- Neck, chest, or abdominal pain
- Vomiting (especially with blood)
- Fever (suggests perforation/infection)
- Blood in stool (black/tarry stool = upper GI bleed)
- Refusal to eat or drink
- Breathing difficulty
Stool checking:
Step 5 - What NOT To Do
| Action | Why to Avoid |
|---|---|
| Do NOT induce vomiting | Brings sharp glass back through esophagus - causes more injury |
| Do NOT give bread/bulky food to "wrap" the glass | No evidence; delays proper assessment |
| Do NOT reassure and discharge without proper exam | Even tiny glass fragments can perforate |
| Do NOT rely on plain X-ray alone to exclude glass | Glass is radiolucent - CT is needed if symptomatic |
Summary Table
| Scenario | Action |
|---|---|
| Asymptomatic, tiny powder particles | Exam + X-ray + observe + return precautions |
| Asymptomatic, larger shard suspected | CT scan + GI/surgical consult |
| Symptomatic, any glass | Urgent endoscopy, surgical consult |
| Signs of perforation (fever, peritonism, free air) | Emergency surgery |
47 yr old male with brethlesness on exertion with hb-6,pcv-23.9,rdw sd 48
Clinical Analysis: 47-Year-Old Male, Exertional Breathlessness
CBC Parameters Interpretation
| Parameter | Patient Value | Normal Range | Interpretation |
|---|---|---|---|
| Hemoglobin | 6 g/dL | 13.5-17.5 g/dL (male) | Severely low - Grade III/IV anemia |
| PCV (Hematocrit) | 23.9% | 40-52% (male) | Severely reduced - consistent with Hb 6 |
| RDW-SD | 48 fL | 35-42 fL | Elevated - significant anisocytosis |
MCV (estimated) = PCV ÷ RBC count × 10. Without RBC count, we use clinical context.
Understanding RDW-SD
| Type | Measures | Normal | Significance |
|---|---|---|---|
| RDW-CV (%) | Coefficient of variation | 11.5-14.5% | Better for detecting early mixed deficiencies |
| RDW-SD (fL) | Standard deviation of cell volume | 35-42 fL | Directly measures absolute size variation; not influenced by MCV |
Diagnostic Possibilities Based on High RDW-SD
1. Iron Deficiency Anemia (IDA) - Most Likely
"Red cell distribution width (RDW) is often elevated, a measure of anisocytosis. Laboratory diagnosis may be demanding in patients with comorbidities. Classical microcytosis and hypochromia can be absent in elderly patients."
"Anemia develops only in the late stage of iron deficiency... anemia often remains undetected until hemoglobin falls below 8 g/dL."
2. Mixed Deficiency Anemia (Iron + B12/Folate)
3. Anemia of Chronic Disease / Inflammation
4. Thalassemia Trait + Iron Deficiency
5. Sideroblastic Anemia
6. Post-Transfusion / Recent Iron Treatment
Key Laboratory Pattern (From Tietz Laboratory Medicine)
| Lab Test | Iron Deficiency | Anemia of Chronic Disease | Thalassemia |
|---|---|---|---|
| Ferritin | Low (<30 µg/L) | Increased | Normal/Increased |
| TIBC | Increased | Decreased | Normal |
| Serum Iron | Low | Low/Normal | Normal/Increased |
| Transferrin saturation | <10% | Normal/>10% | Normal/Increased |
| Hepcidin | Decreased | Increased | Increased/Decreased |
| Serum Transferrin Receptor | Increased | Normal | Normal/Increased |
| RDW | High | Normal/mildly high | Normal/low |
Investigation Plan
Immediate (Confirm Diagnosis)
| Test | Purpose |
|---|---|
| Full CBC with reticulocyte count | Reticulocyte index - is marrow responding? |
| Peripheral blood smear | Morphology - microcytes, hypochromia, pencil cells, target cells, dimorphic picture |
| MCV (if not already available) | Classify anemia - micro/normo/macrocytic |
| Serum ferritin | Best single test for iron stores (low = IDA) |
| Serum iron + TIBC + transferrin saturation | Confirm iron deficiency pattern |
| Serum B12 and folate | Rule out mixed deficiency (explains high RDW-SD) |
| LFTs, RFTs | Chronic disease states |
| LDH + bilirubin + Coombs test | Hemolysis screen |
To Find the Cause (Critical in Middle-Aged Male)
"Tracing the cause may require imaging and endoscopy, procedures essential in males and postmenopausal females to rule out malignancy or other severe causes of bleeding."
| Test | Looking for |
|---|---|
| Fecal occult blood test (FOBT) | GI blood loss |
| Upper GI endoscopy (OGD) | Peptic ulcer, gastric cancer, esophageal varices |
| Colonoscopy | Colon cancer (most important in 47-yr male), polyps, IBD, angiodysplasia |
| Urine for hemoglobin/haemosiderin | Intravascular hemolysis, hematuria |
| HPLC (Hemoglobin electrophoresis) | Thalassemia, hemoglobinopathy |
| CT abdomen if GI scope inconclusive | Occult malignancy |
Immediate Management
This Patient Needs Treatment Now (Hb 6 g/dL)
| Action | Details |
|---|---|
| Assess hemodynamic stability | Pulse, BP, JVP, signs of cardiac failure from anemia (high-output) |
| ECG | Anemia at Hb <7 can cause ischemic changes, tachycardia |
| Blood transfusion | Hb 6 with symptoms = transfusion indicated. Target Hb 8-10 g/dL. Use packed RBCs, 1 unit at a time if cardiac compromise |
| IV iron | Preferred over oral at Hb ≤8 g/dL - rapid repletion. Ferric carboxymaltose or iron sucrose |
| Oral iron | If mild-moderate, alternate-day iron sulfate 200 mg + vitamin C (orange juice) maximizes absorption. Minimum 3 months |
| Do NOT start iron | Until cause of deficiency is identified (or simultaneously investigate) |
| Refer for endoscopy | GI bleeding must be excluded urgently in a 47-yr male with Hb 6 |
Clinical Summary
47-yr male + Hb 6 + PCV 23.9 + RDW-SD 48 fL + exertional dyspnea
│
Severe anemia with significant anisocytosis
│
Most likely: Iron Deficiency Anemia
(from chronic GI blood loss until proven otherwise)
│
Also consider: Mixed IDA + B12/folate deficiency
Dimorphic / sideroblastic anemia
│
KEY INVESTIGATION: Ferritin, peripheral smear, B12, endoscopy
│
KEY CONCERN: Exclude GI malignancy (colon/gastric cancer)
│
TREATMENT: IV iron + transfusion if symptomatic + find and treat cause
37yr old obese with diabetes and dyslipidemia and grade 2 fatty liver taking metformin and glimipramide plans to do weight reduction and shifting to semaglutide
semaglutide NASH nonalcoholic steatohepatitis fatty liver
PMID: 40305708
PMID: 37717295
semaglutide switching from glimepiride metformin T2DM obese NAFLD dose titration 2025
Clinical Management: 37-Year-Old Obese Male with T2DM, Dyslipidemia, Grade 2 Fatty Liver - Shifting to Semaglutide
Why Semaglutide Is the Ideal Choice Here
| Condition | Semaglutide Benefit |
|---|---|
| Obesity | 10-15% body weight reduction (STEP-2 trial) |
| Type 2 Diabetes | HbA1c reduction ~1.4-1.6%; glucose-dependent insulin secretion (no hypoglycemia) |
| Dyslipidemia | Reduces LDL, triglycerides, VLDL |
| Grade 2 Fatty Liver (NAFLD/MASH) | Directly proven to resolve steatohepatitis and reduce fibrosis |
The Evidence Base
For NAFLD/MASH (Strongest Recent Evidence)
In 1197 patients with biopsy-proven MASH + fibrosis stage 2-3, semaglutide 2.4 mg/week:
- Resolution of steatohepatitis: 62.9% vs 34.3% placebo (p<0.001)
- Reduction in fibrosis: 36.8% vs 22.4% placebo (p<0.001)
- Mean body weight loss: -10.5% vs -2.0% placebo
Across 8 studies (n=2413), semaglutide significantly reduced:
- ALT by 14 U/L, AST by 6.9 U/L
- Liver fat content by 4.97%
- Liver stiffness by 0.96 kPa
- Improved HbA1c and lipid profile
For Obesity + T2DM
The Medication Transition Plan
What to Stop and Why
| Drug | Decision | Reason |
|---|---|---|
| Glimepiride - STOP | Discontinue when starting semaglutide | Sulfonylureas cause weight gain (counteracts goal), hypoglycemia risk (especially as semaglutide improves glycemia), no cardiovascular benefit. Lippincott Pharmacology: "Hypoglycemia most common with this class. Weight gain can occur." |
| Metformin - CONTINUE | Keep ongoing | First-line; weight neutral; proven CV/mortality benefit; synergistic with semaglutide. Meta-analysis confirms semaglutide + metformin combination significantly improves glycemic control, weight, BMI, and lipids vs semaglutide alone |
Semaglutide Dosing - Two Options Based on Goal
| Indication | Formulation | Starting Dose | Titration | Target Dose |
|---|---|---|---|---|
| T2DM only | Ozempic (SC weekly) | 0.25 mg/week x 4 weeks | 0.5 mg x 4 weeks → 1 mg x 4 weeks | 1 mg/week |
| Obesity + T2DM (this patient) | Wegovy (SC weekly) | 0.25 mg/week x 4 weeks | 0.5 → 1.0 → 1.7 → 2.4 mg (4 weeks each step) | 2.4 mg/week |
Injection Technique
- Subcutaneous injection into abdomen, thigh, or upper arm
- Same day each week, any time of day, with or without food
- Rotate injection sites
Practical Transition Protocol
Week 1-4
- Stop glimepiride on Day 1 of semaglutide initiation (or taper over 1 week if high HbA1c and concerned about glucose control)
- Start semaglutide 0.25 mg SC once weekly
- Continue metformin at current dose
- Monitor blood glucose more frequently for first 2 weeks
Week 5-8
- Increase semaglutide to 0.5 mg/week
- Monitor HbA1c at 8 weeks
Week 9-12 onward
- Increase to 1 mg → 1.7 mg → 2.4 mg (4 weeks at each step)
- Slow titration reduces GI side effects
Monitoring Schedule
| Parameter | Frequency |
|---|---|
| Blood glucose / HbA1c | 8 weeks, then 3-monthly |
| Fasting lipid panel | 3 monthly initially |
| Liver enzymes (ALT, AST) | 3 monthly - expect improvement |
| LFT + liver ultrasound | 6 months (document NAFLD regression) |
| Body weight / BMI / waist circumference | Monthly |
| Renal function (for metformin monitoring) | 6 monthly |
| Thyroid (TSH) | Baseline - semaglutide carries theoretical thyroid C-cell risk (rodent data) |
Managing the Fatty Liver (Grade 2 NAFLD)
- Semaglutide - now the most evidence-backed pharmacotherapy for NAFLD/MASH
- Weight loss 5-10% alone can reduce liver fat by 40-80% - semaglutide helps achieve this
- Stop alcohol completely if any intake
- Statin use is safe and beneficial in NAFLD + dyslipidemia - do NOT avoid statins for fear of liver toxicity (LFTs rarely worsen with statins in NAFLD)
- Monitor: Repeat ultrasound and LFTs at 6 months to document regression
- Consider FibroScan (transient elastography) to baseline liver stiffness and monitor for fibrosis
Managing Dyslipidemia
- High triglycerides
- Low HDL
- High small-dense LDL
- Atorvastatin 10-20 mg or Rosuvastatin 10 mg
- T2DM is a high CV risk state - statin is indicated
- Semaglutide itself reduces triglycerides and LDL, complementing statin therapy
Side Effects to Counsel the Patient On
| Side Effect | How to Manage |
|---|---|
| Nausea (most common - 20-44%) | Eat smaller meals, avoid fatty/spicy food, take with water; usually resolves by week 8 |
| Vomiting/diarrhea | Slow titration minimizes this |
| Constipation | Increase hydration and fiber |
| Pancreatitis (rare) | Stop if persistent severe abdominal pain; not suitable if personal/family history of pancreatitis |
| Gallstones | Rapid weight loss increases cholelithiasis risk - warn patient about RUQ pain |
| Injection site reactions | Mild, usually self-limiting |
Absolute Contraindications
- Personal or family history of medullary thyroid carcinoma
- Multiple Endocrine Neoplasia syndrome type 2 (MEN2)
Lifestyle - The Non-Negotiable Foundation
"With at least 5% and more so with 10% weight reduction, improvement in insulin sensitivity results in favorable modifications in many components of metabolic syndrome."
| Component | Target |
|---|---|
| Caloric deficit | 500 kcal/day below maintenance = ~0.5 kg/week loss |
| Physical activity | 150-300 min/week moderate intensity (brisk walking, cycling) |
| Diet pattern | Mediterranean or low-carb diet; reduce saturated fat and refined carbohydrates |
| Alcohol | Complete abstinence (worsens NAFLD) |
| Sleep | Screen for obstructive sleep apnoea (common in obese T2DM) |
Summary Management Plan
37M - Obese + T2DM + Dyslipidemia + Grade 2 NAFLD
Currently: Metformin + Glimepiride
CHANGES:
✓ STOP Glimepiride (weight gain, hypoglycemia, no CV benefit)
✓ CONTINUE Metformin
✓ START Semaglutide 0.25mg SC weekly → titrate to 2.4mg
✓ ADD Statin (atorvastatin/rosuvastatin) for dyslipidemia
✓ Lifestyle: 500 kcal deficit + 150 min exercise/week
✓ Monitor: HbA1c, LFTs, lipids, weight, liver ultrasound at 6 months
EXPECTED OUTCOMES at 6-12 months:
- Weight: -10-15% body weight
- HbA1c: -1.4-1.6%
- Liver fat: significant regression of Grade 2 NAFLD
- Lipids: improved TG, LDL, HDL
- Cardiovascular risk: reduced
What about semaglutide oral preparations
oral semaglutide type 2 diabetes weight loss efficacy
oral semaglutide Rybelsus vs injectable dose comparison efficacy weight loss NAFLD 2024 2025
PMID: 37385278
PMID: 38874130
PMID: 37385279
Oral Semaglutide - Complete Clinical Guide
The Pharmaceutical Innovation: How It Works
- SNAC is a fatty acid derivative that acts as an absorption enhancer
- It creates a microenvironment of locally high pH around the tablet in the stomach, protecting semaglutide from acid degradation
- SNAC also facilitates transcellular absorption through the gastric mucosa
- Result: semaglutide is absorbed directly through the stomach wall (not the intestine)
Currently Available Oral Semaglutide Formulations
1. Rybelsus (Oral Semaglutide for T2DM) - FDA approved 2019
| Feature | Details |
|---|---|
| Indication | Type 2 Diabetes (not approved for obesity alone) |
| Doses available | 3 mg, 7 mg, 14 mg tablets |
| Frequency | Once daily |
| Starting dose | 3 mg/day x 30 days (tolerability dose, minimal glycemic effect) |
| Titration | 3 mg → 7 mg → 14 mg (minimum 30 days at each step) |
| Maximum approved dose | 14 mg/day |
| HbA1c reduction | 14 mg: -1.08 to -1.4% vs placebo |
| Weight loss | 14 mg: ~3-4 kg over 26-52 weeks |
- Take on an empty stomach first thing in the morning
- Swallow whole with max 120 mL (half glass) plain water ONLY
- Wait at least 30 minutes before eating, drinking anything other than water, or taking other medications
- Do NOT crush or split tablet
2. Oral Wegovy (High-Dose Oral Semaglutide for Obesity) - FDA approved December 2025
| Feature | Details |
|---|---|
| Indication | Obesity (BMI ≥30) or overweight (BMI ≥27) with comorbidity |
| Doses available | Up to 25 mg/day |
| Titration | Slow escalation over weeks |
| Weight loss (OASIS 4 trial) | -13.6% body weight at 64 weeks |
Head-to-Head: Oral vs Injectable Semaglutide
| Parameter | Rybelsus 14 mg (oral) | Ozempic 1 mg (injectable) | Wegovy 2.4 mg (injectable) | Oral Semaglutide 50 mg (OASIS) |
|---|---|---|---|---|
| Route | Daily oral | Weekly SC injection | Weekly SC injection | Daily oral |
| HbA1c reduction | ~1.1-1.4% | ~1.5-1.8% | ~1.6% | ~2.0% |
| Weight loss | ~3-4 kg (~4.2%) | ~5-7 kg (~10%) | ~15% body weight | ~15.1% body weight |
| Bioavailability | ~1% | ~89% | ~89% | ~1% but higher dose compensates |
| Convenience | Daily, strict fasting rules | Once weekly injection | Once weekly injection | Daily, strict fasting rules |
| CV outcome data | PIONEER-6 (non-inferior) | SUSTAIN-6 (superior) | Awaited | Awaited |
The PIONEER and OASIS Trial Evidence
PIONEER PLUS Trial (Lancet 2023 - PMID 37385279)
| Dose | HbA1c reduction | Body weight change |
|---|---|---|
| 14 mg | -1.5% | Reference |
| 25 mg | -1.8% (p=0.0006 vs 14 mg) | Greater loss |
| 50 mg | -2.0% (p<0.0001 vs 14 mg) | Greatest loss |
OASIS 1 Trial (Lancet 2023 - PMID 37385278)
- Mean weight loss: -15.1% at 68 weeks vs -2.4% placebo
- 85% of participants lost ≥5% body weight
- 54% lost ≥15% body weight
- This matches injectable Wegovy 2.4 mg performance
The conclusion: At 50 mg oral dose, weight loss equals the injectable 2.4 mg subcutaneous formulation.
Meta-analysis (Zhang et al., J Clin Pharmacol 2024 - PMID 38874130)
- HbA1c reduction: dose-dependent (3 mg: -0.61%, 7 mg: -1.12%, 14 mg: -1.08% vs placebo)
- Oral semaglutide outperformed sitagliptin, empagliflozin, liraglutide, and dulaglutide head-to-head
- Less hypoglycemia vs positive comparators (no sulfonylurea-level risk)
- GI side effects higher than placebo but manageable
For This Patient (37M, Obese + T2DM + Fatty Liver)
Should he use oral or injectable semaglutide?
| Factor | Oral (Rybelsus/Oral Wegovy) | Injectable (Ozempic/Wegovy) |
|---|---|---|
| Needle phobia | ✅ Better | ❌ |
| Convenience | ❌ Daily + strict fasting rules | ✅ Once weekly, any time |
| Weight loss (matched dose) | ✅ Comparable at 50 mg | ✅ Best with 2.4 mg |
| NAFLD evidence | Indirect (via weight loss + ALT reduction) | Direct RCT evidence (NEJM 2025 Phase 3 trial used injectable 2.4 mg) |
| Compliance risk | ⚠️ High - strict fasting required | ✅ More flexible |
| Availability | Rybelsus widely available; Oral Wegovy still rolling out | Widely available |
| Cost | Similar | Similar |
- Injectable semaglutide 2.4 mg (Wegovy) is preferred - it has direct Phase 3 MASH/NAFLD evidence (NEJM 2025), better real-world compliance, and the highest weight loss data
- Oral semaglutide (Rybelsus 14 mg) is an acceptable alternative if patient refuses injections or has needle phobia - continue with metformin; titrate to 14 mg maximum
- Oral semaglutide 50 mg (if available) achieves comparable weight loss to injectable Wegovy and could be considered for obesity indication
Oral Semaglutide Dosing for T2DM (Rybelsus)
Month 1: 3 mg once daily (tolerability - minimal glucose effect)
Month 2: 7 mg once daily (therapeutic)
Month 3+: 14 mg once daily (maximum approved dose for T2DM)
Taking rules:
- Empty stomach on waking
- ≤120 mL water only
- Wait 30 min before anything else
- If missed: skip and resume next day (do NOT double dose)
Key Side Effects of Oral Semaglutide
| Side Effect | Incidence | Management |
|---|---|---|
| Nausea | ~20-30% | Improves with titration; eat small meals |
| Diarrhea | ~10-15% | Usually transient |
| Vomiting | ~10% | Slow titration; antiemetics if needed |
| Constipation | ~5-10% | Hydration, fibre |
| Decreased appetite | Common (desired effect) | |
| Pancreatitis (rare) | <1% | Stop if severe abdominal pain |
Summary: Oral vs Injectable for This Patient
ORAL SEMAGLUTIDE (Rybelsus)
├── Available NOW for T2DM
├── 14 mg maximum dose
├── -1.1% HbA1c, ~4 kg weight loss
├── Good for: needle-averse patients
└── Limitation: less weight loss at approved T2DM dose; strict fasting rules
ORAL SEMAGLUTIDE 50 mg (new formulation)
├── Matches injectable Wegovy weight loss (-15%)
├── For obesity indication
└── Still rolling out globally (FDA approved Dec 2025)
INJECTABLE SEMAGLUTIDE 2.4 mg (Wegovy) ← PREFERRED FOR THIS PATIENT
├── Best NAFLD/MASH evidence (NEJM Phase 3 2025)
├── -15% weight loss, -10.5% in MASH trial
├── Once weekly - best adherence
└── Gold standard for obesity + T2DM + NAFLD