Glycogen storage disorders investigations
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive information from multiple authoritative textbooks. Let me compile the complete answer.
Glycogen Storage Disorders — Investigations
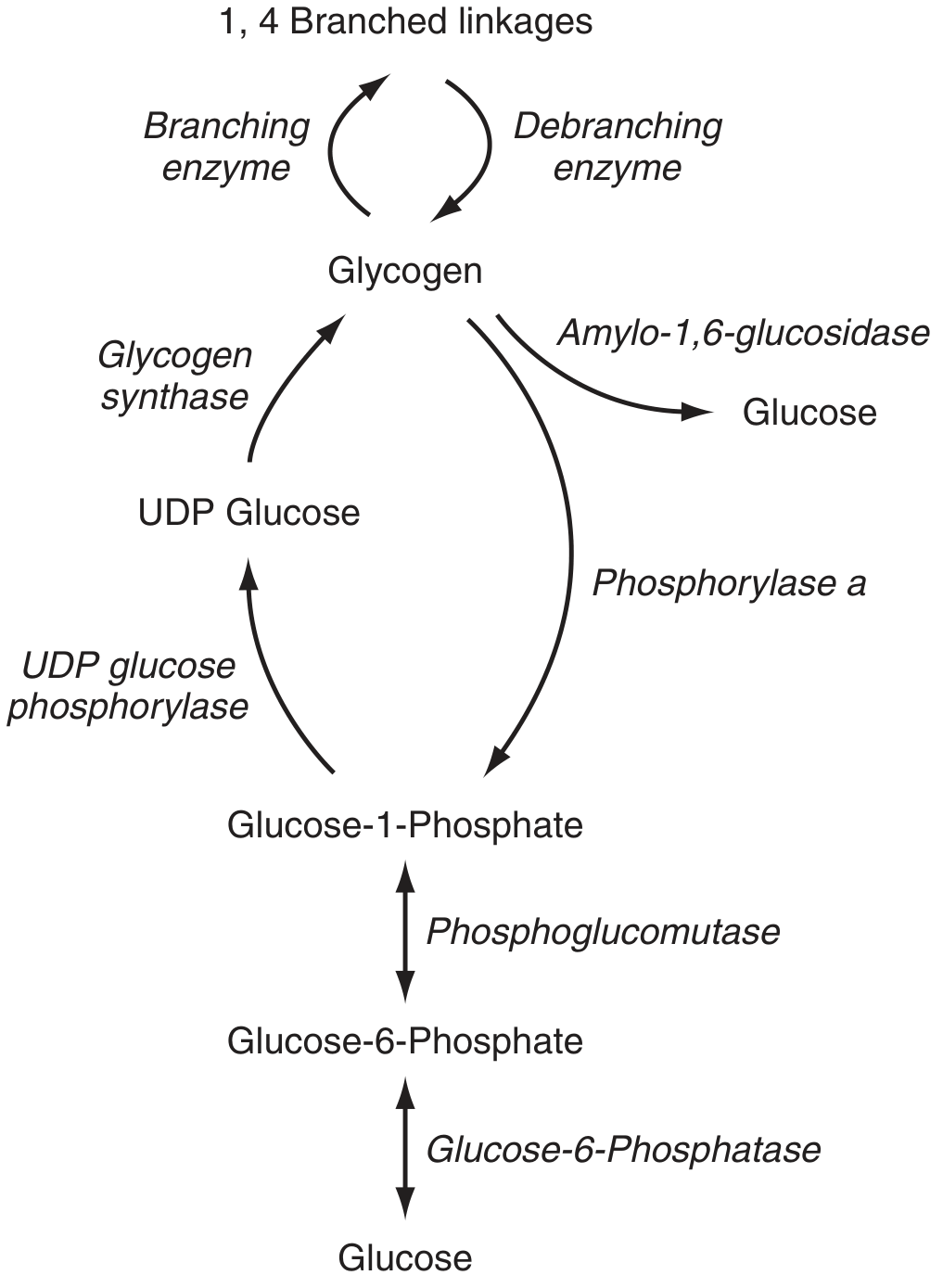
Overview
Glycogen storage diseases (GSDs) are ≥12 inherited disorders of glycogen metabolism. Collectively they occur in ~1:10,000–25,000 births. Because liver and skeletal muscle have the highest glycogen turnover, these are the tissues most affected. Investigation follows a tiered approach: biochemical screening → metabolic provocation tests → enzyme assay → molecular genetics.
First-Line Biochemical Investigations (All Types)
| Test | Significance |
|---|---|
| Fasting plasma glucose | Hypoglycemia is hallmark of hepatic GSDs (GSD I, III, VI, IX); blood glucose 20–50 mg/dL tolerated without symptoms because ketones/lactate serve as alternative CNS fuel |
| Serum lactate | Markedly elevated in GSD I (↑↑↑); normal or mildly elevated in GSD III; absent lactic acidosis helps distinguish GSD III from GSD I |
| Uric acid | Hyperuricemia in GSD I (lactate competitively inhibits renal tubular urate secretion + increased uric acid synthesis) |
| Triglycerides / lipid profile | Hypertriglyceridaemia in GSD I (↑↑↑ lipolysis due to low glucose); also elevated in GSD III |
| Liver transaminases (ALT/AST) | Markedly elevated in GSD III (and GSD IV); helps differentiate from GSD I (where transaminases are normal/mildly raised) |
| Creatine kinase (CK) | Elevated at rest in GSD III (myopathic form), V (McArdle), and VII (Tarui); key marker of muscle involvement |
| Insulin / glucagon ratio | Low insulin and raised glucagon during hypoglycemia |
| Ketones | Ketonaemia/ketonuria during hypoglycaemia — present in GSD III/VI/IX; absent or low in GSD I |
Provocation / Dynamic Tests
Glucagon stimulation test
- Administer glucagon (0.02–0.03 mg/kg IV/IM); measure glucose at 0, 15, 30, 45, 60 min
- Normal: glucose rises ≥30 mg/dL
- GSD I: no rise in blood glucose; lactate rises markedly
- GSD III, VI, IX: blunted or absent rise after prolonged fasting, but partial response post-prandially (distinguishes from GSD I)
Galactose tolerance test (oral or IV)
- Galactose → glucose-1-phosphate → glucose-6-phosphate → glucose (via glucose-6-phosphatase)
- GSD I: failure of blood glucose to rise after galactose administration is diagnostic (glucose-6-phosphatase cannot complete the final step); lactate rises instead
- Still used to confirm GSD I before enzyme assay
Epinephrine tolerance test (historical)
- IM epinephrine: normal response = blood glucose rises 35–45 mg/dL within 40–60 min
- GSD I: no glucose rise; GSD III: no rise after fasting, partial rise after carbohydrate feeding
- Rarely used today; replaced by molecular testing
Ischaemic forearm exercise test (McArdle / GSD V & VII)
- Patient squeezes hand dynamometer 30× under tourniquet (ischaemic conditions)
- Blood sampled for lactate and ammonia before and 1, 2, 3, 5, 10 min after
- Normal: both lactate and ammonia rise (2–5× baseline lactate)
- GSD V (McArdle) / GSD VII (Tarui): ammonia rises normally, but lactate fails to rise (blocked glycolysis in muscle)
- Caution: the non-ischaemic (aerobic) version is safer and preferred now
Enzyme Assays (Tissue-Specific)
The specific diagnosis is confirmed by demonstrating the enzyme defect in relevant tissue:
| GSD Type | Enzyme Deficiency | Diagnostic Tissue |
|---|---|---|
| I (von Gierke) | Glucose-6-phosphatase | Liver biopsy (intact vs. disrupted microsomes differentiates Ia from Ib) |
| II (Pompe) | Acid α-glucosidase (GAA) | Dried blood spot (DBS) for newborn screening; confirmed by leukocytes, muscle, or fibroblasts |
| III (Cori/Forbes) | Glycogen debranching enzyme (amylo-1,6-glucosidase) | Liver + muscle biopsy; erythrocytes (occasionally) |
| IV (Andersen) | Branching enzyme | Leukocytes, cultured fibroblasts, liver/muscle biopsy |
| V (McArdle) | Muscle phosphorylase | Muscle biopsy (histochemistry + enzymatic) |
| VI (Hers) | Liver phosphorylase | Liver biopsy; RBCs or WBCs |
| VII (Tarui) | Muscle phosphofructokinase | Muscle biopsy; erythrocytes (also show partial enzyme deficiency → haemolysis) |
| IX | Phosphorylase kinase | RBCs/WBCs (for X-linked form); liver/muscle biopsy |
Molecular Genetic Testing
Now the primary recommended method of diagnosis (Goldman-Cecil):
- Gene-targeted testing: single-gene sequencing or multigene panel (clinically preferred first-line where genotype is strongly suspected)
- Comprehensive testing: whole-exome sequencing (WES) or whole-genome sequencing (WGS) when panel testing is non-diagnostic
- Common mutations are population-specific (e.g. GSD Ia: p.Arg83Cys in Caucasian/Jewish; c.648G→T splicing in Japanese; GSD Ib: c.1042_1043delCT in Caucasian)
- Used for prenatal diagnosis and carrier detection
Tissue Biopsy
| Biopsy Finding | Significance |
|---|---|
| Liver biopsy — light microscopy with PAS/diastase | Massive glycogen accumulation (PAS+, diastase-sensitive); fat accumulation in GSD I; fibrosis/cirrhosis in GSD III and IV |
| Liver biopsy — electron microscopy | Monoparticulate glycogen (GSD I, III, VI) vs. abnormal polyglucosan/amylopectin-like material (GSD IV) |
| Muscle biopsy — ATPase, phosphorylase histochemistry | Absent phosphorylase staining in GSD V; sub-sarcolemmal vacuoles in GSD II (lysosomal glycogen) |
| Muscle biopsy — EM | Lysosomal glycogen accumulation characteristic of GSD II (Pompe) |
Imaging
| Modality | Findings |
|---|---|
| Ultrasound abdomen | Hepatomegaly (bright, echogenic liver); renomegaly in GSD I; splenomegaly in GSD III/IV |
| MRI liver | Increased glycogen content; distinguishes steatosis (GSD I) from fibrosis |
| Echocardiography | Hypertrophic cardiomyopathy in GSD II (Pompe) — critical for management; also GSD III |
| ECG | Short PR interval and massive QRS voltages in infantile Pompe; conduction defects in GSD III |
| Skeletal X-ray/bone densitometry | Osteoporosis in GSD I (long-term) |
Newborn Screening (GSD II — Pompe)
- Dried blood spot (DBS) acid α-glucosidase (GAA) enzyme activity assay — several countries include GSD II in neonatal screening
- Confirmed by GAA gene sequencing ± lymphocyte/fibroblast enzyme assay
- Rationale: enzyme replacement therapy (ERT) with alglucosidase alfa is most effective when started before symptom onset
Type-Specific Investigation Highlights
GSD I (von Gierke) — Key Lab Fingerprint
- Hypoglycaemia + lactic acidosis + hyperuricaemia + hypertriglyceridaemia + hepatomegaly
- No glucose response to glucagon or galactose
- Confirm: liver biopsy → G6Pase assay (intact microsomes: Ia; + disrupted microsomes required: Ib); G6PC1 / SLC37A4 sequencing
GSD II (Pompe) — Key Lab Fingerprint
- Infantile: cardiomegaly, hypotonia, elevated CK, very high GAA deficiency on DBS
- Late-onset: progressive proximal myopathy, respiratory failure, CK elevated, normal/mild CK in some
- Confirm: DBS GAA activity → GAA gene sequencing; muscle biopsy shows lysosomal glycogen
GSD III (Cori/Forbes) — Distinguishing from GSD I
- Elevated transaminases (GSD I has normal AST/ALT), elevated CK (muscle involvement in IIIa)
- Hyperglycaemic response to galactose (unlike GSD I)
- Lower uric acid and lactate than GSD I
- Confirm: debranching enzyme (AGL) assay in erythrocytes or liver/muscle; AGL gene sequencing
GSD IV (Andersen) — Key Findings
- Hepatosplenomegaly → cirrhosis → liver failure
- Abnormal glycogen (amylopectin-like) on biopsy (PAS+, poorly diastase-digestible)
- Confirm: branching enzyme (GBE1) assay in leukocytes/fibroblasts; GBE1 sequencing
GSD V (McArdle) — Key Lab Fingerprint
- Elevated resting CK; myoglobinuria after exercise
- Ischaemic forearm test: ammonia rises, lactate fails to rise
- "Second-wind" phenomenon
- Confirm: absent phosphorylase on muscle biopsy histochemistry; PYGM sequencing
GSD VII (Tarui) — Key Lab Fingerprint
- Like GSD V but also haemolytic anaemia (RBCs share the PFK defect)
- Hyperbilirubinaemia + reticulocytosis + pigmenturia
- Glucose does not relieve symptoms (unlike GSD V)
- Confirm: PFK assay in RBCs + muscle biopsy; PFKM sequencing
Summary Algorithm
Suspected GSD
│
├── Hepatic features (hepatomegaly, hypoglycaemia)
│ ├── + Lactic acidosis + ↑↑ uric acid + ↑↑ TG → GSD I → galactose test → liver biopsy/G6PC gene
│ ├── + ↑ transaminases + ↑ CK ± cardiomyopathy → GSD III → AGL enzyme/gene
│ ├── + Cirrhosis/liver failure → GSD IV → GBE enzyme/gene
│ └── Mild, asymptomatic hepatomegaly → GSD VI / IX → phosphorylase/kinase assay
│
└── Muscle features (exercise intolerance, cramps, rhabdomyolysis)
├── + Cardiomyopathy + hypotonia (infant) → GSD II → DBS GAA assay
├── + Forearm test: ↑↑ NH₃, flat lactate + "second wind" → GSD V → PYGM gene
└── + Haemolytic anaemia + flat lactate forearm test → GSD VII → PFK assay
Sources:
- Tietz Textbook of Laboratory Medicine, 7th Edition — Table 35.1, GSD types I–VII
- Henry's Clinical Diagnosis and Management by Laboratory Methods — Tables 17.6 & 17.7
- Goldman-Cecil Medicine (International Edition) — Chapter 191 (Kishnani)
- Sleisenger & Fordtran's GI and Liver Disease — Chapter 77
This is a shared conversation. Sign in to Orris to start your own chat.