Multicystic disease
Reading File
Asking for Preferences
Finding Sources
Searching PubMed
"multicystic dysplastic kidney"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Finding Sources
Searching Images
multicystic dysplastic kidney ultrasound gross pathology
I now have comprehensive information from multiple authoritative textbooks plus imaging. Let me compile the full answer.
Multicystic Dysplastic Kidney (MCDK)
Definition & Overview
MCDK is a severe, non-inherited (sporadic) form of renal dysplasia resulting from abnormal nephrogenesis — most likely from in utero ureteral obstruction occurring very early in fetal development. It is the second most common cause of an abdominal mass in newborns, after hydronephrosis due to pelvi-ureteric junction obstruction (PUJO).
- Incidence: 1 in 1,000–4,000 live births (unilateral)
- Bilateral MCDK is incompatible with life (→ Potter sequence)
Pathology
- The kidney is non-reniform and non-functional
- Contains a "bunch of grapes" / "cluster of grapes" appearance — multiple non-communicating cysts of varying sizes with no identifiable renal parenchyma
- The kidney lacks a well-formed collecting system or pelvicalyceal architecture
- Loose mesenchyme, sometimes containing cartilage, fills the space between cysts
- Can involve the entire kidney or, rarely, just a segment
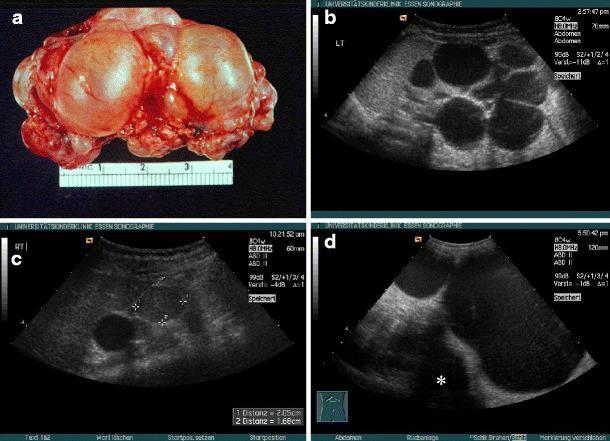
MCDK: (a) gross specimen showing cluster of cysts; (b–d) ultrasound variants including classic non-communicating cysts, giant MCDK crossing midline, and one with a superimposed nephroblastoma.
Diagnosis
Prenatal
- Detectable on antenatal ultrasound as early as 15 weeks' gestation
- Multiple cysts evident; may be confused with severe P3 urinary tract dilation (UTD)
- Other reniform phenotypes (not classic) have been described
Postnatal
| Modality | Findings |
|---|---|
| Renal ultrasound | Multiple non-communicating cysts of varying size; paucity of intervening solid tissue; no identifiable pelvis |
| DMSA renal scintigraphy | Photopenic (non-functional) area in the renal fossa with background surrounding activity |
| MRI | Large multicystic kidney with varied non-communicating cysts (useful when US equivocal) |
| VCUG | Historically used to screen for VUR (found in ~17–23%); routine use now questioned |
Associated Anomalies
Contralateral urinary tract abnormalities occur in ~25% of cases, including:
- Vesicoureteral reflux (VUR) — ~20%
- Ureteropelvic junction obstruction (UPJO)
- Contralateral kidney hypoplasia, malrotation, or ectopia
Bilateral MCDK — Potter Sequence
Bilateral involvement causes Potter syndrome (oligohydramnios sequence):
- Widely separated eyes with epicanthal folds
- Broad nasal bridge
- Low-set ears
- Receding chin
- Pulmonary hypoplasia → incompatible with life
Natural History & Complications
| Feature | Detail |
|---|---|
| Involution | Progressive reduction in kidney size; 60% contract by age 2; 20–25% are undetectable by US within 2 years |
| Hypertension | Rare complication |
| UTI | Rare; prophylactic antibiotics generally not recommended |
| Malignant transformation | Wilms tumor and RCC described, but incidence not significantly different from the general population |
| Compensatory hypertrophy | Contralateral kidney undergoes expected hypertrophy |
Management
Surveillance (Current Standard)
- Renal/bladder ultrasound in the early postnatal period to confirm prenatal findings
- Renal ultrasound every 3 months for the first year, then every 6 months up to involution or at least age 5
- VCUG and DMSA: recommended with the caveat that this is not evidence-based; withholding these studies is also acceptable practice
- Prophylactic antibiotics: generally not recommended (low UTI risk)
Monitoring for Contralateral Kidney
In the setting of a functional solitary kidney:
- Blood pressure measurement
- Assessment for proteinuria
- Consideration of yearly ultrasound through puberty to monitor for hyperfiltration injury
Nephrectomy Indications
- Routine nephrectomy is no longer recommended (previous concern for Wilms tumor not substantiated by systematic review)
- Consider surgical removal if:
- Increase in cyst size (unusual — warrants malignancy exclusion)
- Persistent cystic kidney with a solid component
- Hypertension is present and thought to be related
Key Differentials
| Condition | Distinguishing Feature |
|---|---|
| Hydronephrosis (PUJO) | Cysts communicate; central large cyst (dilated pelvis); some residual parenchyma |
| ADPKD/ARPKD | Bilateral; family history; reniform shape preserved; hereditary |
| Cystic nephroma | Well-encapsulated; typically in young boys; herniation into ureter |
| Glomerulocystic kidney disease | Dilation of Bowman's capsule; more related to ADPKD genotype |
Sources: Campbell Walsh Wein Urology, p. 494 | National Kidney Foundation Primer on Kidney Diseases, 8e | Bailey and Love's Short Practice of Surgery, 28e | Quick Compendium of Clinical Pathology, 5e
This is a shared conversation. Sign in to Orris to start your own chat.