Thalassemia pediatric from ghai
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive content to write a thorough answer. Let me compile the full response on Thalassemia in Pediatrics, in the style typically found in Ghai's Essential Pediatrics (which is not in this library), using the best available textbook content.
Note: Ghai's Essential Pediatrics is not in this textbook library. The content below is drawn from Robbins & Kumar Basic Pathology, Robbins, Cotran & Kumar Pathologic Basis of Disease, and Goldman-Cecil Medicine - all authoritative sources covering thalassemia comprehensively.
Thalassemia - Pediatric
Definition & Epidemiology
Thalassemia is a genetically heterogeneous disorder caused by germline mutations that decrease synthesis of either α-globin or β-globin, leading to anemia, tissue hypoxia, and hemolysis from the imbalance in globin chain synthesis.
- Endemic in the Mediterranean basin (thalassa = "sea" in Greek), Middle East, tropical Africa, Indian subcontinent, and Asia
- Among the most common inherited disorders of humans
- Prevalence is maintained because heterozygous carriers are protected against falciparum malaria
- The two α-chains of HbA are encoded by two α-globin genes on chromosome 16; the β-chain by a single β-globin gene on chromosome 11
Classification
β-Thalassemia (most clinically important)
| Clinical Syndrome | Genotype | Clinical Features |
|---|---|---|
| β-Thalassemia Major (Cooley's anemia) | Homozygous (β⁰/β⁰, β⁰/β⁺, β⁺/β⁺) | Severe anemia; regular blood transfusions required |
| β-Thalassemia Intermedia | Variable | Moderately severe anemia; regular transfusions NOT required |
| β-Thalassemia Minor (trait) | Heterozygous (β⁺/β, β⁰/β) | Asymptomatic or mild; red cell abnormalities seen |
α-Thalassemia
| Deleted α-genes | Syndrome | Features |
|---|---|---|
| 1 gene deleted | Silent carrier | No anemia, normal CBC |
| 2 genes deleted | α-Thalassemia trait | Mild microcytic anemia |
| 3 genes deleted | HbH disease | Moderate hemolytic anemia, HbH (β₄ tetramers) |
| 4 genes deleted | Hb Bart's hydrops fetalis | Lethal in utero (γ₄ tetramers, no O₂ delivery) |
Molecular Pathogenesis of β-Thalassemia
More than 100 causative mutations have been identified, mostly point mutations (unlike α-thalassemia which is mainly deletional). Three main classes:
- Splicing mutations - most common cause of β⁺-thalassemia; some create ectopic splice sites within introns
- Promoter region mutations - reduce transcription by 75-80%; result in β⁺ alleles
- Chain terminator mutations (nonsense mutations, frameshifts) - most common cause of β⁰-thalassemia; completely block β-globin translation
Pathophysiology
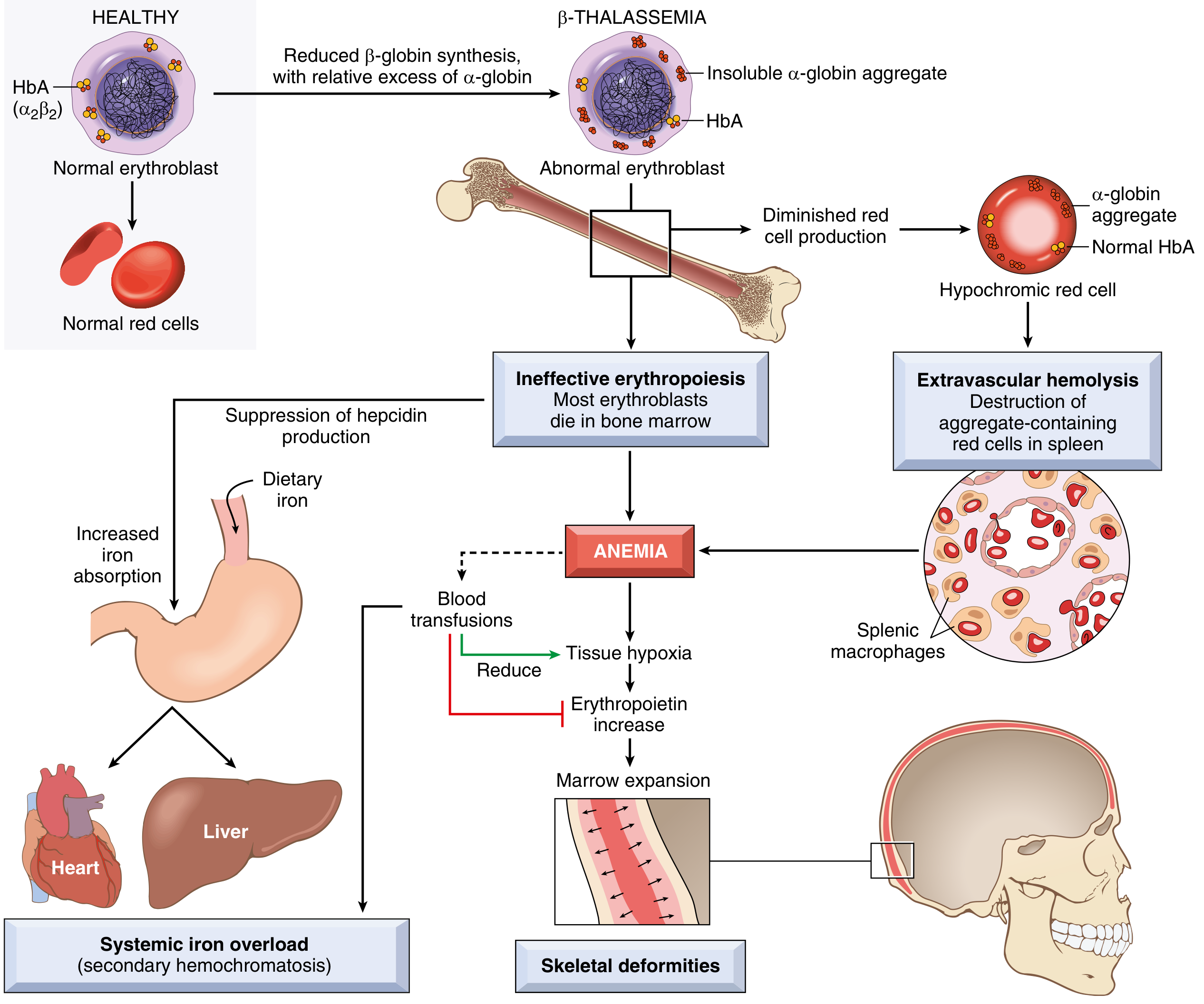
Pathophysiology of β-Thalassemia Major (Robbins & Kumar Basic Pathology)
Anemia arises by two mechanisms:
- Inadequate HbA formation → small (microcytic), poorly hemoglobinized (hypochromic) red cells
- Accumulation of unpaired α-globin chains → toxic precipitates that severely damage red cell and erythroid precursor membranes → apoptosis of erythroid precursors in bone marrow = ineffective erythropoiesis
Consequences of ineffective erythropoiesis:
- Erythropoietin surge → massive marrow expansion → bone deformities (frontal bossing, maxillary hypertrophy = "thalassemic facies")
- Extramedullary hematopoiesis → hepatosplenomegaly, paravertebral masses (sternum, ribs)
- Suppression of hepcidin → increased dietary iron absorption → iron overload (even without transfusions)
Consequences of extravascular hemolysis:
- Aggregate-containing red cells destroyed by splenic macrophages
- Splenomegaly → hypersplenism → pancytopenia, increased transfusion requirement
Clinical Features
β-Thalassemia Major (Cooley's Anemia) - Pediatric Presentation
- Symptoms appear during the first year of life (as γ→β switching occurs after birth)
- Without treatment: hemoglobin cannot be maintained above 5 g/dL
- Pallor, failure to thrive, jaundice, progressive abdominal distension
- Thalassemic facies: frontal bossing, prominent malar eminences, protruding jaw (due to marrow expansion)
- Hepatosplenomegaly - constant finding; leads to pancytopenia
- Growth retardation and delayed puberty in inadequately treated children
- Recurrent infections, spontaneous fractures, gallstones (cholelithiasis), leg ulcers in early childhood
- Skeletal deformities from marrow expansion (hair-on-end appearance on skull X-ray)
Iron Overload Complications (in older children/adolescents)
- Heart failure - multifactorial (chronic anemia + myocardial iron toxicity); leading cause of death
- Liver disease - cirrhosis from iron overload (± hepatitis B/C from transfusions)
- Endocrinopathies - diabetes mellitus, hypothyroidism, hypogonadism (delayed puberty/primary amenorrhea)
- Osteoporosis/osteopenia
β-Thalassemia Minor
- Asymptomatic, or mild microcytic hypochromic anemia
- Commonly mistaken for iron deficiency anemia - do not respond to iron
- Key distinction: elevated HbA₂ (>3.5%) on HPLC/electrophoresis
HbH Disease (3-gene α-thalassemia)
- Moderate hemolytic anemia; Hb usually 8-9 g/dL
- Does not require regular transfusions
- Hemolytic crises during acute infections
- Requires folic acid supplementation (2-5 mg/day), especially in children
Diagnosis
CBC: Microcytic (low MCV), hypochromic (low MCH), low Hb
Peripheral smear: Microcytes, hypochromic cells, target cells, nucleated RBCs, basophilic stippling, Heinz bodies (in HbH)
Hemoglobin electrophoresis / HPLC:
- β-Thal major: absent or markedly reduced HbA, elevated HbF (20-90%), elevated HbA₂
- β-Thal minor: HbA₂ > 3.5% (most reliable marker)
- HbH: HbH band on electrophoresis Serum ferritin & iron studies: Elevated (iron overload) - helps distinguish from iron deficiency DNA analysis: Definitive diagnosis; identifies specific mutations MRI liver/heart: Quantifies iron overload (T2* MRI)
Treatment
Transfusion Therapy
- Goal: Maintain pre-transfusion Hb > 9-10.5 g/dL (suppresses ineffective erythropoiesis, prevents marrow expansion and skeletal deformities)
- Transfusions given every 2-5 weeks
- Use leukoreduced packed red cells to minimize transfusion reactions and pathogen transmission
- Initiation requires: definitive molecular diagnosis + severity of anemia on repeated measurement + clinical criteria (failure to thrive, bone changes)
Iron Chelation Therapy
Started when ferritin > 1000 ng/mL or after ~10-20 transfusions
| Agent | Route | Notes |
|---|---|---|
| Deferoxamine (DFO) | SC/IV infusion (8-12 hrs/night, 5-7 days/week) | Gold standard; requires compliance; audiometry/ophthalmology monitoring |
| Deferasirox | Oral, once daily | Convenient; renal function monitoring required |
| Deferiprone | Oral, TID | Best for cardiac iron; agranulocytosis risk |
Splenectomy
- Indicated when annual blood consumption increases progressively (>200 mL/kg/year of packed RBCs) and is responsible for significant iron accumulation despite good chelation, OR symptomatic splenomegaly/hypersplenism
- Delay until child is at least 5 years old (risk of overwhelming post-splenectomy infection ~50% mortality)
- Mandatory vaccinations before splenectomy: pneumococcus, meningococcus, H. influenzae type b
- Post-splenectomy: lifelong penicillin prophylaxis + increased thrombosis risk
Curative Options
- Allogeneic bone marrow/stem cell transplantation - best results in pediatric patients with HLA-identical sibling donors (Goldman-Cecil, 2024); adults have more iron overload and worse outcomes
- Gene therapy - corrective gene therapy now approved as safe and effective for β-thalassemia; increasingly available (Harrison's Principles, 2025)
Newer Therapies
- Luspatercept (recombinant fusion protein, activin receptor type IIB ligand trap) - approved for adults with transfusion-dependent β-thalassemia; reduces transfusion burden by ≥33%; given SC 1.0-1.25 mg/kg every 3 weeks
- Hydroxyurea - can increase HbF levels; more benefit in sickle cell disease than thalassemia
- α-globin expression reduction - experimental approach for HbE β-thalassemia
Prevention & Genetic Counseling
- Carrier screening (CBC: low MCV/MCH → confirm with HPLC) for at-risk ethnic populations
- Both parents must be carriers for a child to have thalassemia major
- Prenatal diagnosis possible by chorionic villus sampling (CVS) at 10-12 weeks or amniocentesis
- Preimplantation genetic diagnosis (PGD) available
- Carriers (thalassemia minor) should receive appropriate genetic counseling
- During pregnancy, carrier mothers may become more anemic - monitor in 2nd/3rd trimesters + folic acid supplementation
Key Points for Exams
| Feature | β-Thal Major | β-Thal Minor | α-Thal Trait |
|---|---|---|---|
| Presentation | 1st year of life | Asymptomatic | Asymptomatic |
| Anemia | Severe (<5 g/dL) | Mild | Mild |
| HbA₂ | Elevated | >3.5% | Normal |
| HbF | Markedly elevated | Normal/slight ↑ | Normal |
| Transfusion needed | Yes | No | No |
| Classic X-ray | Hair-on-end skull | Normal | Normal |
- Robbins & Kumar Basic Pathology, pp. 387-390
- Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 602-607
- Goldman-Cecil Medicine, Chapter 148, pp. 1710-1715
Nephtotic syndrome in pediatrics
Reading File
You've run out of free credits. Sign up for a free account to continue using Orris.
This is a shared conversation. Sign in to Orris to start your own chat.