Idiopathic intracranial hypertension
Reading File
Finding Sources
Searching PubMed
"idiopathic intracranial hypertension"[MeSH Terms]
Reading File
Reading File
Searching PubMed
"pseudotumor cerebri" OR "idiopathic intracranial hypertension" AND treatment
Reading File
Reading File
Searching Images
idiopathic intracranial hypertension papilledema fundus MRI
I now have comprehensive content. Let me compile the full answer.
Idiopathic Intracranial Hypertension (IIH)
Synonyms: Pseudotumor cerebri, benign intracranial hypertension
Definition
IIH is a condition of increased intracranial pressure (ICP) with no identifiable structural or metabolic cause. It manifests classically with papilledema, headache, and elevated CSF opening pressure, yet normal brain parenchyma and CSF composition.
Epidemiology
IIH is almost exclusively a disease of obese women of reproductive age:
- Mean age: ~29 years
- 97.6% female
- Mean BMI: ~39.9 kg/m²
- Incidence rises sharply with increasing obesity
— Bradley and Daroff's Neurology in Clinical Practice
Pathophysiology
The exact mechanism remains debated. Proposed mechanisms include:
- Impaired CSF absorption at arachnoid granulations (possibly due to adipokines or hormonal influences on CSF secretion/resorption)
- Venous outflow obstruction: Bilateral transverse sinus stenosis is found in nearly all IIH patients. Whether it is the cause or consequence is debated — a self-perpetuating cycle may exist where elevated ICP compresses the sinuses → venous outflow obstruction → further ICP rise
- Hormonal/metabolic factors: Obesity, sex steroids, vitamin A metabolism disorders, and glucocorticoid dysregulation have all been implicated
Clinical Features
From the IIH Treatment Trial (IIHTT):
| Symptom | Frequency |
|---|---|
| Headache | 84% |
| Transient visual obscurations | 68% |
| Back pain | 53% |
| Pulse-synchronous tinnitus | 52% |
| Visual loss | 32% |
| Diplopia (lateral rectus palsy / CN VI) | less common |
- Headache is the most common presenting symptom; most common phenotypes are migraine (52%) and tension-type (22%)
- Transient visual obscurations result directly from papilledema and raised ICP
- Diplopia results from CN VI (lateral rectus) palsy — a false localizing sign from raised ICP
- Pulsatile/pulse-synchronous tinnitus is characteristic
Diagnostic Criteria
(Modified Friedman/Mollan criteria)
IIH:
| Criterion |
|---|
| A. Papilledema |
| B. Normal neurological exam (except CN VI palsy) |
| C. Normal brain MRI/CT; venous thrombosis excluded |
| D. Normal CSF composition |
| E. LP opening pressure >25 cm H₂O (≥250 mm H₂O in adults) |
IIH Without Papilledema (IIHWOP): Criteria B–E plus unilateral or bilateral CN VI palsy.
Pressure must be measured in the lateral decubitus position. Sitting pressures are invalid. LP opening pressure ≥250 mm CSF is required.
Workup
- MRI brain with contrast — mandatory before LP to exclude intracranial mass, hydrocephalus
- MR Venography (MRV) — to exclude cerebral venous sinus thrombosis (key IIH mimic) and assess for bilateral transverse sinus stenosis
- Lumbar puncture with manometry — CSF should be clear, colorless, with normal protein/glucose/cells; high opening pressure confirms diagnosis
- Ophthalmologic evaluation — confirm papilledema (distinguish from pseudopapilledema/drusen), assess visual fields and acuity
MRI findings suggestive of IIH:
- Partially empty sella
- Bilateral optic nerve sheath distension
- Flattening of posterior sclera
- Bilateral transverse sinus flow gaps/stenosis on MRV
Imaging
Multimodal imaging findings in IIH:
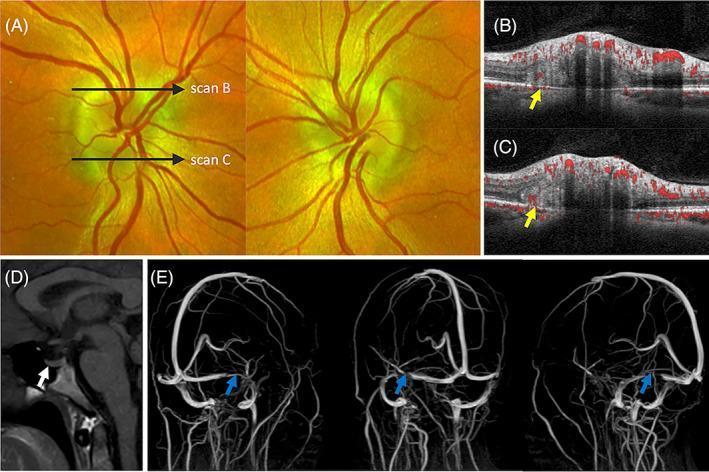
Bilateral papilledema on fundoscopy, OCT with PHOMS, MRI showing empty sella and patulous optic nerve sheaths (white arrow), and MRV showing bilateral transverse sinus flow gaps (blue arrows)
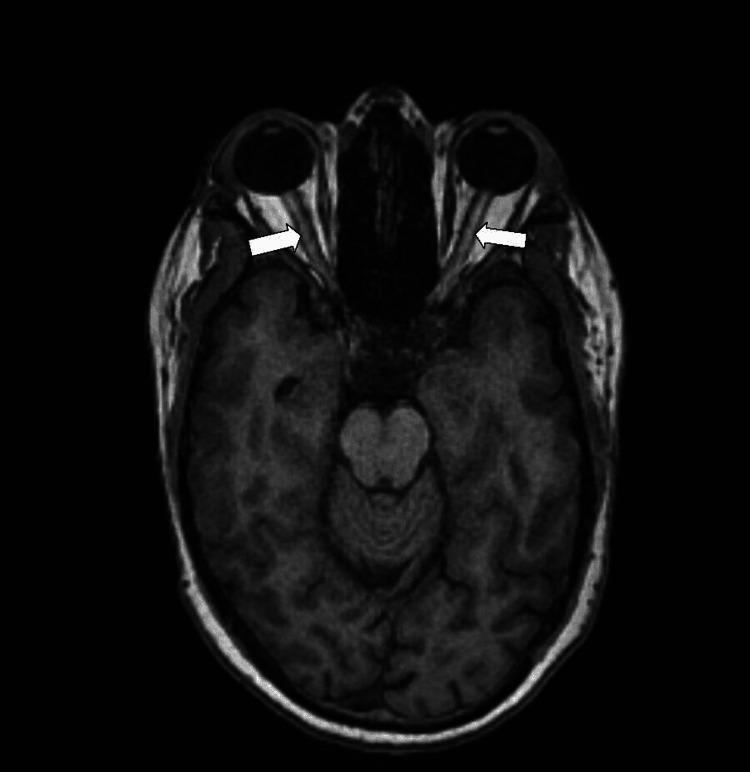
Axial T1 MRI of orbits: bilateral optic nerve sheath distension (arrows) — a hallmark of raised ICP
Secondary Causes ("Pseudotumor Cerebri") — Must Exclude
| Category | Examples |
|---|---|
| Cerebral venous abnormalities | Sinus thrombosis, bilateral jugular ligation, middle ear infection, AV fistulas, SVC syndrome, prior SAH/meningitis |
| Medications & exposures | Tetracyclines (tetracycline, minocycline, doxycycline), vitamin A & retinoids (isotretinoin, all-trans retinoic acid), anabolic steroids, human growth hormone, corticosteroid withdrawal, lithium, levonorgestrel |
| Endocrine/medical conditions | Addison disease, hypoparathyroidism, hypercapnia, sleep apnea, Pickwickian syndrome, anemia, renal failure, Turner/Down syndrome |
Management
Primary goal: Prevent permanent visual field loss.
1. Conservative
- Weight loss: 5–10% body weight reduction can normalize ICP and resolve papilledema
2. Medical
- Acetazolamide (carbonic anhydrase inhibitor — reduces CSF production): First-line drug
- Start: 500 mg twice daily
- Titrate up as tolerated; doses up to 4 g/day supported by IIHTT evidence
- The IIHTT showed acetazolamide + weight loss improves papilledema, ICP, and quality of life
- Topiramate: Alternative/adjunct (also promotes weight loss)
- Other diuretics: Furosemide used as adjunct
- Headache management: Migraine-specific therapies (triptans, etc.) based on phenotype
3. Surgical / Procedural
Indicated when vision is threatened despite maximum medical therapy:
| Procedure | Indication |
|---|---|
| Optic nerve sheath fenestration (ONSF) | Progressive visual loss — decompress optic nerve directly |
| CSF diversion shunt (LP shunt or VP shunt) | Refractory raised ICP with visual threat |
| Venous sinus stenting | Selected patients with documented focal transverse sinus stenosis + significant pressure gradient on transvenous manometry |
The venous sinus stenting approach is supported by evidence that a pressure gradient across a stenosis can be corrected endovascularly (mean venous pressure improved from 51 → 17 mmHg in one series).
4. Lumbar puncture
- Serial LPs can provide temporary relief (headache + visual obscurations)
- Used as a bridge to definitive therapy, especially in pregnancy
IIH in Pregnancy
- Typically worsens with pregnancy; most commonly develops around 14th gestational week
- Visual loss in ~10–20%
- MRV is mandatory to exclude cerebral venous thrombosis
- Management:
- Diet modification (avoid ketosis)
- Serial LPs preferred over medications
- Acetazolamide is controversial in pregnancy; most guidelines suggest avoiding in the first trimester; restrict its use to after 20 weeks when clinically essential
- Corticosteroids (dexamethasone/prednisone) for 2-week courses when vision is threatened
- ONSF or lumboperitoneal shunting if vision fails despite serial LPs
- Vaginal delivery with epidural analgesia is generally preferred
Prognosis
- Most patients improve with weight loss ± acetazolamide
- Permanent vision loss can occur in untreated or refractory cases — visual fields (especially nasal inferior) are at greatest risk
- Risk of recurrence if weight is regained
- In pregnancy, IIH typically resolves postpartum; recurrence in subsequent pregnancies is unusual
Key References
- Bradley and Daroff's Neurology in Clinical Practice, Chapter 102
- IIHTT (IIH Treatment Trial) — Friedman et al., 2014; Smith and Friedman, 2017
- Mollan et al., J Neurol Neurosurg Psychiatry 2018 (consensus guidelines)
- Friedman et al., Neurology 2013 (revised diagnostic criteria)
- Yiangou et al., Nat Rev Neurol 2023 — recent review on disease mechanisms (PMID: 37957260)
- Friedman DI, Neurol Clin 2024 — updated clinical review of pseudotumor cerebri syndrome (PMID: 38575259)
Pseudobulbar vs bulbar palsy
Finding Sources
Searching Images
pseudobulbar palsy bulbar palsy comparison upper lower motor neuron
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
I have comprehensive content from multiple authoritative textbooks. Here is the full comparison:
Pseudobulbar Palsy vs Bulbar Palsy
Core Concept
The bulbar muscles — muscles of the jaw, face, tongue, pharynx, and larynx — are controlled by motor nuclei in the lower brainstem (CN V, VII, IX, X, XI, XII). These nuclei themselves receive supranuclear (corticobulbar) input from the motor cortex.
| Bulbar Palsy | Pseudobulbar Palsy | |
|---|---|---|
| Neuron type | Lower motor neuron (LMN) | Upper motor neuron (UMN) |
| Site of lesion | Motor nuclei of CN IX–XII in the brainstem (bulb = medulla) | Bilateral corticobulbar tracts (supranuclear) |
| Laterality | Can be unilateral or bilateral | Must be bilateral |
| Why "pseudo"? | The prefix pseudo- distinguishes it from true (LMN) bulbar palsy — the clinical syndrome mimics it but the underlying neuron type differs |
Clinical Features Comparison
| Feature | Bulbar Palsy (LMN) | Pseudobulbar Palsy (UMN) |
|---|---|---|
| Dysarthria | Nasal, flaccid ("hot potato" speech) — difficulty with lingual (r, n, l), labial (b, m, p), palatal (k, g) consonants | Spastic, strained, slow, effortful — "Donald Duck" quality |
| Dysphagia | Present — food lodges between cheek and teeth; liquids regurgitate nasally | Present, generally milder |
| Tongue | Wasted, fasciculating, lies flaccid on floor of mouth | Small, spastic, slow-moving — no wasting or fasciculations |
| Fasciculations | Present (tongue, facial muscles) | Absent |
| Muscle atrophy | Present — tongue shrivels; lower facial muscles sag | Absent |
| Jaw jerk | Absent or depressed | Brisk / exaggerated (pathognomonic UMN sign) |
| Gag reflex | Absent or reduced | Hyperactive |
| Palatal movement | Impaired or absent | Impaired |
| Emotional lability | Absent | Present — pathological (forced/spasmodic) laughing and crying (pseudobulbar affect) — the defining UMN feature |
| Primitive reflexes | Absent | Snout reflex, suck reflex present |
| Limb signs | LMN signs if motor neuron disease | UMN signs (spasticity, hyperreflexia, Babinski) in limbs |
| Tongue clonus | Absent | May be present |
| "Bulldog" reflex | May occur in ALS (mixed) | Present in severe cases |
Pseudobulbar Affect (Emotional Lability)
This is the hallmark distinguishing feature of pseudobulbar palsy:
- Spontaneous, unmotivated, or disproportionate laughing and crying
- Does not reflect the patient's actual emotional state
- Results from bilateral corticobulbar tract damage releasing brainstem emotional expression circuits from cortical inhibition
- Often causes significant embarrassment to the patient
"Spontaneous or unmotivated crying and laughter uniquely characterize pseudobulbar palsy." — Bradley and Daroff's Neurology in Clinical Practice
Causes
Bulbar Palsy (LMN)
Diseases affecting the motor nuclei of CN IX–XII or their axons:
- Motor neuron disease (progressive bulbar palsy — the most common cause)
- Guillain-Barré syndrome (acute onset, no atrophy)
- Poliomyelitis
- Diphtheria
- Syringobulbia
- Brainstem tumors / infarcts (if targeting motor nuclei)
- Myasthenia gravis (NMJ — mimics bulbar palsy)
- Kennedy disease (spinobulbar muscular atrophy — X-linked LMN)
- Fazio-Londe disease (childhood progressive bulbar palsy)
Pseudobulbar Palsy (UMN)
Diseases causing bilateral corticobulbar tract damage:
- Bilateral lacunar infarcts (most common — "lacunar state")
- ALS (mixed UMN + LMN — unique situation where both coexist)
- Multiple sclerosis
- Diffuse hypoxic-ischemic encephalopathy
- Progressive supranuclear palsy (PSP)
- Wilson disease
- Bilateral MCA territory strokes
- Cerebral vasculitis
"The most common pathologic bases are lacunar infarctions or other cerebrovascular lesions, diffuse hypoxic-ischemic encephalopathy, ALS, and multiple sclerosis." — Adams and Victor's Principles of Neurology, 12th Ed.
Special Case: ALS (Mixed Bulbar + Pseudobulbar)
ALS is the only common clinical situation where both spastic (pseudobulbar) and atrophic (true bulbar) palsy coexist. This mixed picture — fasciculating, wasted tongue alongside a brisk jaw jerk and emotional lability — is highly characteristic.
Neuroanatomical Pathway Diagram
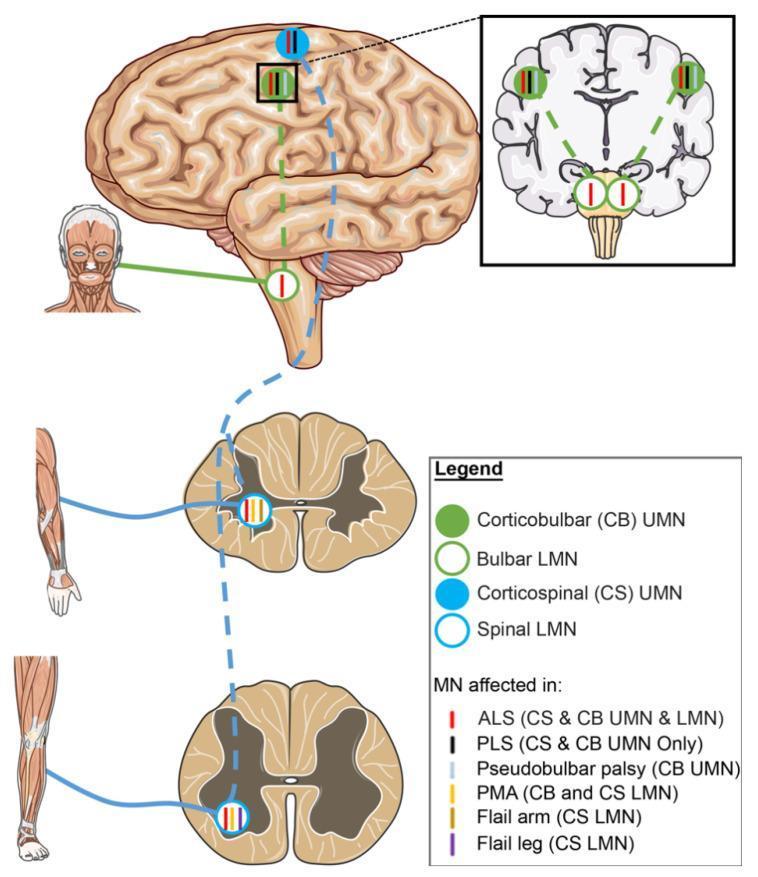
Schematic showing UMN (solid circles) and LMN (open circles) in the corticobulbar (green) and corticospinal (blue) tracts. Pseudobulbar palsy = corticobulbar UMN involvement only; bulbar palsy = LMN involvement at brainstem nuclei.
Quick Memory Summary
| Bulbar | Pseudobulbar | |
|---|---|---|
| Neuron | LMN | UMN |
| Tongue | Flaccid, Fasciculating, Fatigued (wasted) | Spastic, Small, Slow |
| Jaw jerk | ↓ / absent | ↑ brisk |
| Emotional lability | ✗ | ✓ (pathognomonic) |
| Gag reflex | ↓ | ↑ |
| Atrophy | ✓ | ✗ |
| Cause | MND, GBS, polio | Lacunar strokes, ALS, MS |
Sources: Bradley and Daroff's Neurology in Clinical Practice; Adams and Victor's Principles of Neurology, 12th Ed.; Harrison's Principles of Internal Medicine 22E; Goldman-Cecil Medicine
JNE MYopathy
Finding Sources
Searching the Web
JNE myopathy juvenile neuromuscular
Finding Sources
Finding Sources
Searching PubMed
"GNE myopathy"[MeSH Terms] OR "Nonaka myopathy"
Reading File
Reading File
Searching Images
GNE myopathy Nonaka distal myopathy rimmed vacuoles histology muscle biopsy
Loading Image
Finding Sources
I have comprehensive content. Here is the full answer:
Note: "JNE myopathy" appears to be a typographic variant of GNE Myopathy. The answer below covers GNE myopathy in full.
GNE Myopathy
Synonyms: Nonaka myopathy · Autosomal recessive hereditary inclusion body myopathy (hIBM2) · Distal myopathy with rimmed vacuoles (DMRV)
Definition
GNE myopathy is a rare, autosomal recessive distal myopathy caused by loss-of-function mutations in the GNE gene, which encodes the bifunctional enzyme UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase (GNE/MNK) — the rate-limiting enzyme in the biosynthesis of sialic acid. Reduced sialic acid sialylation of muscle glycoproteins is the proposed pathomechanism.
Genetics
| Feature | Detail |
|---|---|
| Gene | GNE |
| Locus | Chromosome 9p13-q1 |
| Inheritance | Autosomal recessive |
| Protein | UDP-N-acetylglucosamine-2-epimerase / N-acetylmannosamine kinase |
| Function | Rate-limiting step in sialic acid biosynthesis |
| Common founder mutations | p.Met712Thr (Middle Eastern/Persian Jewish — Udaka mutation); p.Val572Leu (Japanese) |
Epidemiology
- Onset: second to third decade (late teens to twenties)
- Affects all ethnicities, but over-represented in Persian Jewish and Japanese populations
- Ultra-rare globally (~250–350 families described worldwide in earlier reports, but prevalence rising with genetic diagnosis)
Clinical Features
Onset & Pattern
- Initial weakness: anterior tibial muscles → progressive foot drop and steppage gait
- Extensor forearm muscles also affected early
- Weakness is distal-predominant, with characteristic quadriceps sparing — the hallmark that distinguishes GNE myopathy from most other myopathies
- Upper limb involvement: wrist and finger extensors, intrinsic hand muscles
Progression
- Slow but inexorably progressive
- Ambulation typically lost within 10–15 years of onset
- Ultimately all limb muscles except quadriceps become involved
- Respiratory and cardiac muscles are usually spared (important distinction from many other dystrophies)
Quadriceps Sparing
This is pathognomonic — even when other proximal muscles are severely wasted, the quadriceps remain relatively preserved until very late stages. The reason is unknown but may relate to differential sialylation demands.
Clinical Photograph
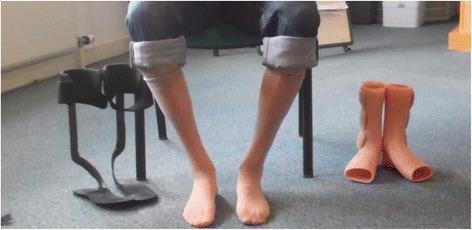
Marked bilateral anterior compartment atrophy with bilateral foot drop and ankle-foot orthoses (AFO) in a patient with GNE myopathy
Laboratory Features
| Test | Finding |
|---|---|
| Serum CK | Mildly elevated — 3–10× normal |
| EMG | Myopathic pattern; early recruitment |
| Nerve conduction studies | Normal |
| Muscle biopsy (LM) | Dystrophic features + rimmed vacuoles (Gomori trichrome) |
| Electron microscopy | 15–18 nm tubulofilamentous inclusions (identical to sporadic IBM inclusions) |
| Inflammation | Absent (unlike sporadic IBM — important distinction) |
| Protein accumulation | Ubiquitin-positive inclusions; TDP-43 inclusions |
Key Biopsy Point
Rimmed vacuoles + tubulofilaments on EM but no significant inflammatory infiltrate = GNE myopathy. Sporadic IBM has the same inclusions but WITH inflammation and is a completely different entity (late-onset, sporadic, immune-mediated component).
MRI Findings
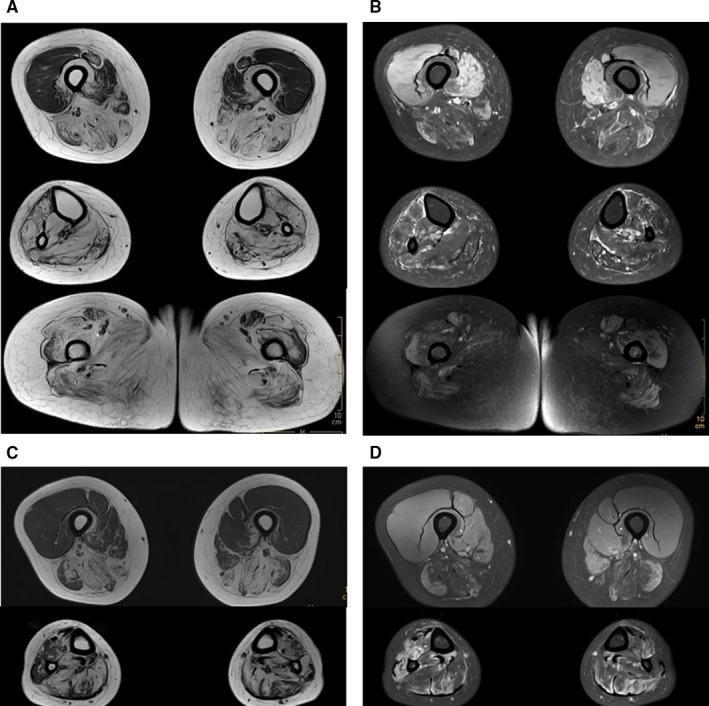
Axial MRI lower limbs in GNE myopathy: T1 (A, C) shows extensive fatty replacement in posterior/medial thigh compartments and lower legs (tibialis anterior, gastrocnemius). PDw SPAIR (B, D) shows muscle edema. Note relative quadriceps preservation.
Distal Myopathies Comparison Table
(From Harrison's Principles of Internal Medicine, 22nd Ed.)
The table below summarizes GNE myopathy alongside other distal myopathies:
| Disease | Onset | Initial Weakness | CK | Inheritance | Gene |
|---|---|---|---|---|---|
| Welander | 5th decade | Hands/wrist extensors | 2–3× | AD | TIA1 |
| Udd (tibial) | 4th–8th decade | Anterior tibial | 2–4× | AD | Titin |
| Markesbery-Griggs | 4th–8th decade | Anterior tibial + distal arms | Mildly ↑ | AD | ZASP |
| Laing | Childhood/early adult | Anterior tibial + neck flexors | Normal/↑ | AD | MYH7 |
| GNE myopathy | 2nd–3rd decade | Anterior tibial; quadriceps spared | 3–10× | AR | GNE |
| Miyoshi | 2nd–3rd decade | Gastrocnemius (posterior) | 20–100× | AR | Dysferlin |
Pathophysiology
The GNE enzyme catalyzes two sequential steps in sialic acid (N-acetylneuraminic acid / NANA) production:
- Epimerase domain: UDP-GlcNAc → ManNAc
- Kinase domain: ManNAc → ManNAc-6-phosphate
Loss of function → ↓ sialic acid → hyposialylation of muscle glycoproteins (e.g., α-dystroglycan, NCAM, integrin) → membrane instability and sarcolemmal dysfunction.
Mutations are distributed across both domains. The homozygous p.Met712Thr (kinase domain) is found in ~95% of affected Middle Eastern Jewish patients.
Diagnosis
- Clinical suspicion: Young adult onset distal weakness with foot drop + quadriceps sparing
- CK: Mildly elevated
- EMG: Myopathic
- Muscle biopsy: Rimmed vacuoles + EM tubulofilaments, no inflammation
- Genetic testing: GNE gene sequencing — confirmatory
Treatment
No disease-modifying therapy is currently approved.
| Approach | Details |
|---|---|
| Ankle-foot orthoses (AFOs) | For foot drop — first-line supportive measure |
| Occupational therapy | Hand/wrist function preservation |
| Sialic acid supplementation | Rationale: restore sialic acid; phase 2/3 trials (aceneuramic acid / ManNAc / sialic acid precursors) — failed to meet primary endpoints in pivotal trials |
| Intravenous immunoglobulin (IVIG) | Anecdotal benefit; not standard |
| Gene therapy | Under investigation |
| Substrate replacement | Extended-release sialic acid (aceneuramic acid / Ultragenyx) — pivotal trial (Extend study) did not show significant benefit |
| Respiratory monitoring | Annual pulmonary function tests recommended in advanced disease |
| Genetic counseling | Autosomal recessive; 25% recurrence risk |
Prognosis
- Slowly progressive over decades
- Loss of ambulation: typically 10–15 years after onset
- Respiratory and cardiac complications are rare but should be monitored in late stages
- Lifespan is generally normal
Key Differentials
| Condition | Distinguishing Feature |
|---|---|
| Sporadic IBM | Late onset (>50 yrs), inflammation on biopsy, finger flexor + quadriceps weakness |
| Miyoshi myopathy | Posterior compartment onset (calves), markedly elevated CK, dysferlin deficiency |
| CMT / HMSN | Neuropathic EMG/NCS, sensory involvement |
| Tibial MD (Udd) | AD inheritance, late onset, titin mutation |
| Foot drop from motor neuropathy | Normal CK, neurogenic EMG, NCS abnormal |
Sources: Bradley and Daroff's Neurology in Clinical Practice; Harrison's Principles of Internal Medicine, 22nd Ed.; Adams and Victor's Principles of Neurology, 12th Ed.
This is a shared conversation. Sign in to Orris to start your own chat.