Thalassemia explain
Reading File
Finding Sources
Searching PubMed
thalassemia
Reading File
Reading File
Reading File
Reading File
Searching Images
thalassemia blood smear pathogenesis
Reading File
Loading Image
Here is a comprehensive explanation of thalassemia based on Robbins & Cotran Pathologic Basis of Disease and Robbins Basic Pathology.
Thalassemia
Definition
Thalassemia is a genetically heterogeneous group of inherited disorders caused by germline mutations that decrease the synthesis of either α-globin or β-globin chains, leading to anemia, tissue hypoxia, and red cell hemolysis from the resulting imbalance in globin chain synthesis. The name comes from the Greek thalassa ("sea"), reflecting its prevalence in Mediterranean populations. — Robbins & Cotran Pathologic Basis of Disease, p. 602
Epidemiology
Thalassemia is endemic in the:
- Mediterranean basin
- Middle East
- Tropical Africa
- Indian subcontinent
- Asia
It is among the most common inherited disorders in humans. Its high prevalence is explained by the protection it confers on heterozygous carriers against falciparum malaria — the same selective pressure that drives the distribution of sickle cell disease.
Genetics & Molecular Basis
| Chain | Gene Location | Gene Count |
|---|---|---|
| α-globin (for HbA's 2 α chains) | Chromosome 16 | 4 genes total (2 per chromosome) |
| β-globin (for HbA's 2 β chains) | Chromosome 11 | 1 gene per chromosome |
β-Thalassemia Mutations (mainly point mutations)
Three major classes:
-
Splicing mutations — most common cause of β⁺-thalassemia. Some destroy normal RNA splice junctions (→ β⁰); others create ectopic splice sites within introns (→ β⁺, partial normal mRNA still made).
-
Promoter region mutations — reduce transcription by 75–80%; always → β⁺.
-
Chain terminator mutations — most common cause of β⁰-thalassemia. Nonsense mutations (premature stop codons) or frameshift mutations (small insertions/deletions); no functional β-globin produced at all.
α-Thalassemia Mutations
Caused mainly by gene deletions (in contrast to β-thalassemia's point mutations). Severity is proportional to the number of α-globin genes deleted out of the four total.
Pathogenesis
Anemia arises through two mechanisms:
-
HbA deficiency → hypochromic, microcytic red cells with reduced O₂-carrying capacity.
-
Globin chain imbalance (especially critical in β-thalassemia):
- Excess unpaired α-chains precipitate within red cell precursors → insoluble inclusions
- Inclusions cause membrane damage → apoptosis of red cell precursors = ineffective erythropoiesis (70–85% of precursors lost in severe β-thalassemia)
- Released RBCs with inclusions → splenic sequestration and extravascular hemolysis
Consequences of Severe Ineffective Erythropoiesis
- Massive erythroid hyperplasia in marrow → extramedullary hematopoiesis (liver, spleen, lymph nodes, even thoracic/abdominal masses)
- Bony expansion → cortical erosion, skeletal deformity, classic "crew-cut" appearance on skull X-ray
- Iron overload: Expanded erythroid mass → ↑ erythroferrone secretion → suppresses hepcidin → ↑ gut iron absorption; combined with repeated transfusions → secondary hemochromatosis → cardiac and hepatic injury
Clinical Classification
β-Thalassemia
| Syndrome | Genotype | Severity |
|---|---|---|
| β-Thalassemia major (Cooley's anemia) | β⁰/β⁰, β⁺/β⁰, β⁺/β⁺ (homozygous) | Severe anemia; transfusion-dependent from infancy |
| β-Thalassemia intermedia | Variable | Moderately severe; not regularly transfusion-dependent |
| β-Thalassemia minor (trait) | β/β⁺ or β/β⁰ (heterozygous) | Mild/asymptomatic; microcytosis on CBC |
β-Thalassemia Major Clinical Features:
- Severe anemia appearing 6–9 months after birth (as HbF declines)
- Hepatosplenomegaly (extramedullary hematopoiesis + hemolysis)
- Skeletal changes: frontal bossing, maxillary prominence ("chipmunk facies"), crew-cut skull X-ray
- Growth retardation, cachexia
- Iron overload → cirrhosis, cardiac failure, endocrinopathies
- Without treatment: death in first decade; with regular transfusions + iron chelation, survival into adulthood
α-Thalassemia
| Syndrome | Genes Deleted | Clinical Features |
|---|---|---|
| Silent carrier | 1 of 4 (−/α, α/α) | Asymptomatic; slight microcytosis |
| α-Thalassemia trait | 2 of 4 | Asymptomatic; like β-thal minor; normal HbA₂ |
| HbH disease | 3 of 4 | Moderate anemia; β-globin tetramers (HbH) form; HbH high O₂ affinity → poor O₂ delivery; prone to precipitation → splenomegaly |
| α-Thalassemia major (Hb Bart's) | All 4 | Incompatible with postnatal life without transfusion; γ-chain tetramers (Hb Bart's); hydrops fetalis; death in utero or shortly after birth |
Clinical Images
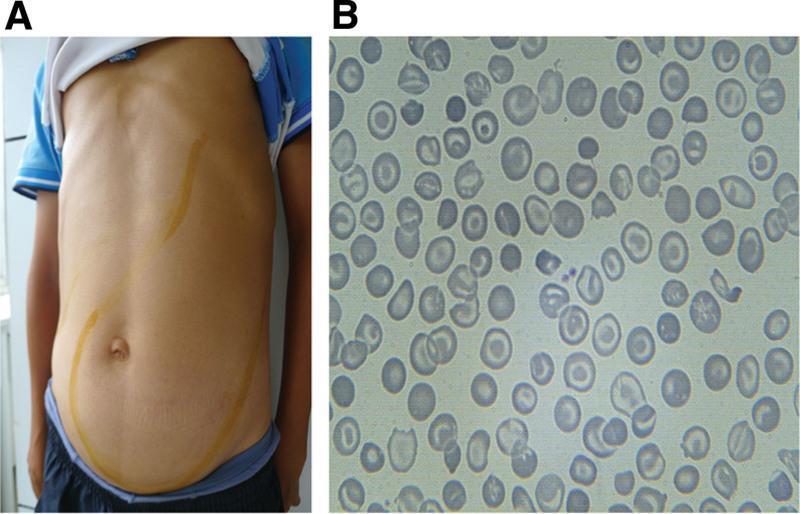
Below: Thalassemia intermedia — Panel A shows massive hepatosplenomegaly (yellow lines demarcating organ borders); Panel B shows the characteristic peripheral blood smear with microcytic, hypochromic RBCs and numerous target cells (codocytes).
Laboratory Findings
| Finding | β-Thal Major | β-Thal Minor |
|---|---|---|
| Hemoglobin | Very low (3–6 g/dL) | Near normal or mildly low |
| MCV/MCH | Markedly reduced | Mildly reduced (microcytosis) |
| Blood smear | Severe poikilocytosis, target cells, nucleated RBCs, basophilic stippling | Mild microcytosis, target cells |
| HbA₂ | Elevated | Elevated (>3.5%) — key diagnostic marker |
| HbF | Markedly elevated | Normal or slightly elevated |
Treatment
| Strategy | Details |
|---|---|
| Regular blood transfusions | Required for thalassemia major; maintains Hb >9–10 g/dL, suppresses ineffective erythropoiesis |
| Iron chelation | Deferoxamine (IV/SC) or deferasirox/deferiprone (oral) — essential to prevent hemochromatosis |
| Splenectomy | For hypersplenism refractory to transfusion increases |
| Hematopoietic stem cell transplantation (HSCT) | Curative; best outcomes in young patients with matched sibling donor |
| Gene therapy | Luspatercept (reduces transfusion burden in β-thal); gene therapy trials (betibeglogene — approved by FDA for β-thal major) |
| HbF inducers | Hydroxyurea increases HbF, benefiting some patients |
Key Summary
- β-Thalassemia = point mutations → ↓ β-globin → excess α-chains → ineffective erythropoiesis + hemolysis
- α-Thalassemia = gene deletions → ↓ α-chains → excess β- or γ-chain tetramers
- Both cause microcytic hypochromic anemia with variable severity
- The central pathological mechanism is globin chain imbalance, not simply hemoglobin deficiency
- Complications include iron overload, skeletal deformity, organomegaly, and growth failure
— Robbins & Cotran Pathologic Basis of Disease, pp. 601–608; Robbins Basic Pathology, pp. 1765–1776
This is a shared conversation. Sign in to Orris to start your own chat.