Wrist drop due to stroke how to differentiate from other causes
wrist drop radial nerve palsy differentiation stroke
wrist drop stroke vs radial nerve palsy differentiation clinical features
Wrist Drop: Differentiating Stroke (Central) from Other Causes
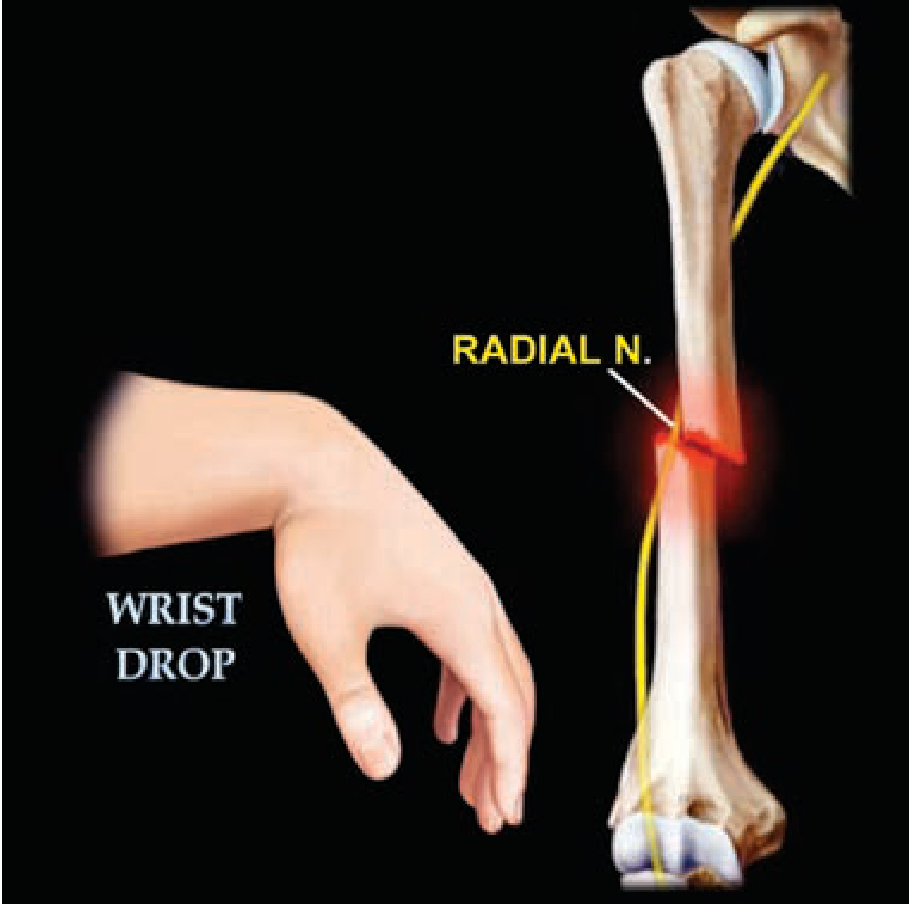
1. Pathophysiology of Each Cause
| Level | Mechanism | Key Point |
|---|---|---|
| Cortical/subcortical stroke | Infarct in hand motor cortex (homunculus, precentral gyrus) or corticospinal tract | Extensors of hand affected preferentially; UMN lesion |
| Radial nerve - axilla | Crutch pressure, humerus fracture at spiral groove | Triceps also affected |
| Radial nerve - spiral groove | "Saturday night palsy," "park bench palsy," humeral shaft fracture | Classic wrist drop; triceps spared |
| Posterior interosseous nerve (PIN) | Arcade of Frohse entrapment, radial tunnel syndrome | Finger drop, NO wrist drop, NO sensory loss |
| C7 radiculopathy | Disc herniation at C6-7 | Wrist extensors + triceps + sensory loss in digits 3-4 |
| Brachial plexus (posterior cord) | Trauma, tumour, neuralgic amyotrophy | Multiple nerve distributions |
2. Core Clinical Differentiation: Stroke vs. Radial Nerve Palsy
Key Bedside Tests
- Ask the patient to make a fist (place the wrist in a neutral position passively)
- Radial nerve palsy: finger flexion strength improves markedly when the wrist is passively supported in neutral, because the flexors (ulnar + median nerve-innervated) are intact
- Stroke: NO significant improvement in the neutral position - extensors AND flexors are all weak because it is a UMN lesion affecting the entire hand
- Radial nerve palsy: Finger flexion (median/ulnar) is PRESERVED - patient can grip strongly
- Stroke: Finger flexion AND abduction are both weak - patient cannot flex OR extend the fingers
- Stroke preferentially damages wrist and finger extensors more than flexors (upper limb corticospinal pattern)
- Radial nerve palsy produces a flaccid wrist/finger drop affecting only radial-innervated muscles; flexors are entirely normal
UMN vs. LMN Signs Table
| Feature | Stroke (UMN) | Radial Nerve Palsy (LMN) |
|---|---|---|
| Tone | Increased (spastic) - may be flaccid acutely | Decreased (flaccid) |
| Reflexes | Hyperreflexia, Babinski present | Reduced/absent triceps, brachioradialis, supinator jerks |
| Muscle wasting | Absent (early) | Present (late, with axonal injury) |
| Fasciculations | Absent | May be present |
| Sensory pattern | No sensory loss OR hemisensory loss (face/arm/leg same side) | Sensory loss confined to radial territory: dorsal first web space, dorsal thumb/index/middle finger |
| Other limbs | May have leg weakness (hemiplegia pattern) | Only the arm affected |
| Face involved? | Yes, if MCA/cortical | No |
| Onset | Sudden (seconds to minutes) | Often positional (waking after sleep, trauma) |
3. Differentiating Peripheral Causes Among Themselves
Radial Nerve by Level (site of injury determines muscles lost)
| Level of Injury | Triceps | Brachioradialis | Wrist Extension | Finger Extension | Sensory Loss |
|---|---|---|---|---|---|
| Axilla (high) | Weak | Weak | Lost | Lost | Posterior arm + dorsal hand |
| Spiral groove | Spared | Weak | Lost | Lost | Dorsal hand/thumb area |
| PIN only (elbow level) | Spared | Spared | Preserved (partial) | Lost | None |
| Superficial radial nerve (wrist) | Normal | Normal | Normal | Normal | Dorsal hand only (pure sensory) |
- THIEME Atlas of Anatomy, p. 389: "Distal radial nerve lesion... no typical wrist drop and no sensory disturbances" (PIN lesion)
- Bradley and Daroff's Neurology, p. 539: "Damage to the radial nerve in the spiral groove results in damage to muscles innervated distally to the triceps. Patients typically present with wrist drop, and sensory symptoms are minimal."
C7 Radiculopathy vs. Radial Nerve Palsy
| Feature | C7 Radiculopathy | Radial Nerve Palsy |
|---|---|---|
| Neck pain/radiation | Common, radiates to arm | Absent |
| Reflexes | Triceps jerk reduced | Triceps (if high) or brachioradialis reduced |
| Sensory | Digits 3 and 4 (ulnar side included) | Dorsal radial hand/web space only |
| Triceps | Weak | Weak only if axillary lesion |
| EMG/NCS | Fibrillations in C7 muscles including serratus anterior, cervical paraspinals | Abnormal only in radial-innervated muscles; paraspinals normal |
| MRI | C6-7 disc herniation | Normal |
4. Additional Causes to Exclude
- Lead neuropathy (lead poisoning): Classic cause of bilateral wrist drop with motor neuropathy; associated with basophilic stippling, anaemia, Burton's lines on gums
- Mononeuritis multiplex (vasculitis, diabetes, leprosy): Multiple single nerve involvement; wrist drop + foot drop in different limbs simultaneously - Robbins Pathologic Basis of Disease: "An affected patient might have a right wrist drop from involvement of the right radial nerve and a left foot drop from peroneal nerve damage. Vasculitis is a common cause..."
- Posterior interosseous nerve (PIN) entrapment: Only finger drop, NO wrist drop, NO sensory loss; lateral elbow pain; occurs at arcade of Frohse
- Humeral shaft fracture: Obvious trauma history; radial nerve in spiral groove injured; assess with X-ray
5. Summary: Practical Approach at Bedside
- Sudden onset + face involvement + leg weakness = Stroke until proven otherwise → MSCT/MRI brain urgently
- Check fist/grip: strong grip = peripheral (radial nerve); weak grip = central
- Check triceps: if weak → lesion is at axilla or C7; if normal → spiral groove or below
- Check sensory: confined to dorsal radial hand = radial nerve; hemisensory = stroke; dermatomal (digits 3-4 + neck pain) = C7 root
- Reflexes: hyperreflexia + Babinski = stroke; hyporeflexia (brachioradialis, triceps) = peripheral
- EMG/NCS: gold standard for peripheral localization; confirms axonal vs. demyelinating, pinpoints level
- Brain MRI (DWI): gold standard for central - cortical hand area ("hand knob") infarct
Rat Kill Poisoning
Rat Kill (Rodenticide) Poisoning
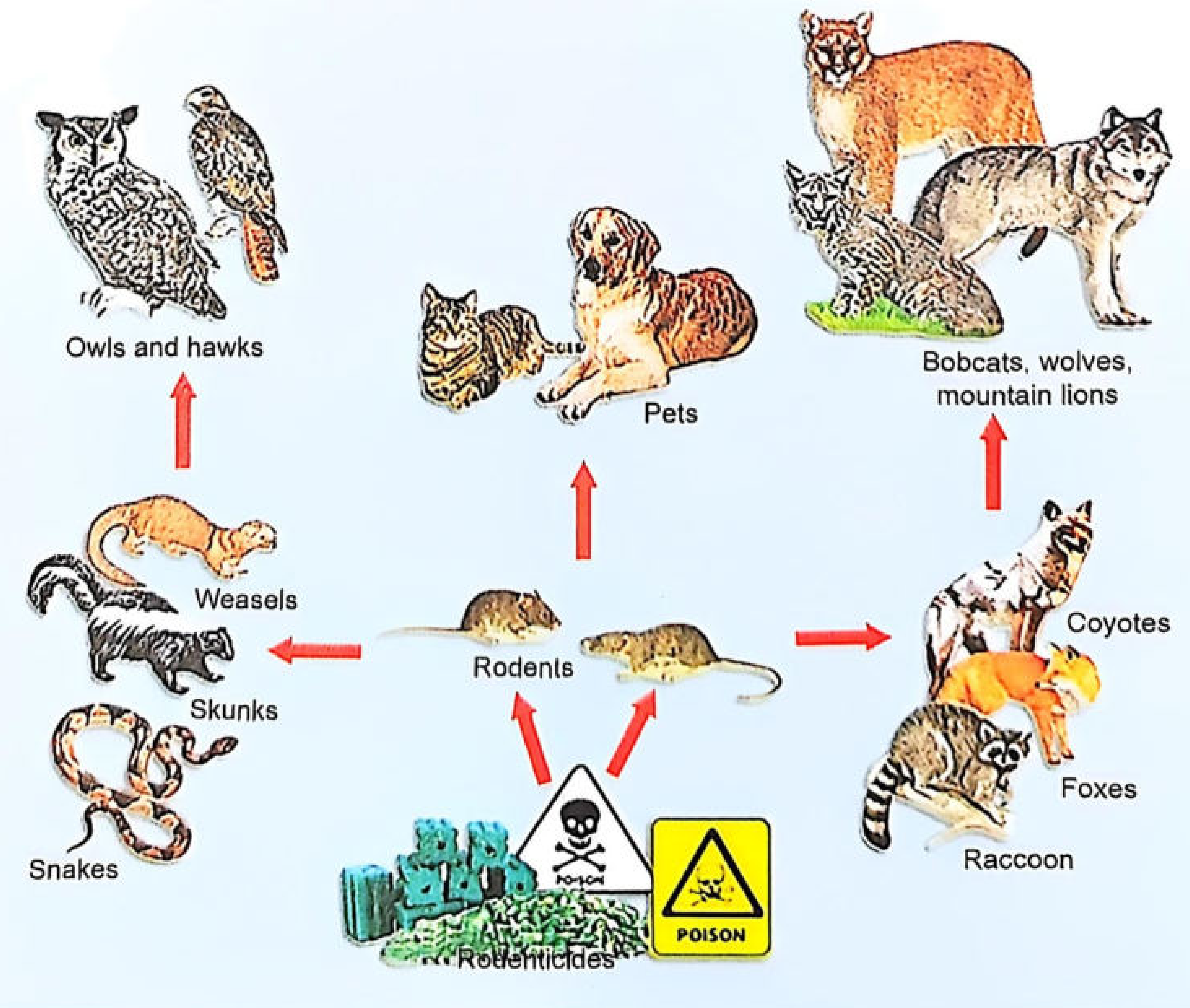
Part 1: Aluminium Phosphide (ALP) Poisoning
Mechanism of Action
- Inhibits the electron transport chain - specifically cytochrome oxidase
- Is a systemic mitochondrial poison affecting every organ
- Rapidly absorbed from GIT by simple diffusion
- Also absorbed via inhalation; excreted via lungs and urine as hypophosphite
- Some ALP is absorbed and metabolized in the liver, releasing phosphine slowly - accounting for prolonged symptoms
Fatal Dose and Period
- Fatal dose: 1-3 g (1-3 tablets); inhalation of 400-600 ppm fatal within 1 hour
- Fatal period: 6-12 hours typically; majority die within 24 hours
- Mortality: 35-100%
Clinical Features
| System | Features |
|---|---|
| GIT | Nausea, vomiting, diarrhea, retrosternal pain |
| CVS | Hypotension, shock, arrhythmias, myocarditis, pericarditis, acute congestive heart failure |
| Respiratory | Cough, dyspnea, cyanosis, pulmonary edema, ARDS, respiratory failure |
| Hepatic | Jaundice, hepatitis, hepatomegaly |
| Renal | Acute renal failure |
| CNS | Headache, dizziness, altered mental state, restlessness, convulsions, coma |
| Rare | Muscle wasting, bleeding diathesis (widespread capillary damage) |
Chemical Test for Diagnosis
- Mix 5 mL gastric aspirate + 15 mL water in a flask → cover with 0.1N silver nitrate-impregnated filter paper → heat at 50°C for 15-20 min → paper turns black if PH₃ present
- Patient breathes through AgNO₃-impregnated paper mask for 15-20 min → turns black (positive only if >6 g ALP ingested)
Postmortem Findings
- Garlic-like odor at mouth, nostrils, gastric contents
- Liver, spleen, kidneys, and brain: congested
- Centrizonal hemorrhagic necrosis of liver
- Histopathology: Stomach - congestion, edema, leucocytic infiltration, sloughing of gastric mucosa; Lungs - congestion, edema, lymphocytic infiltration; Kidneys - tubular degeneration; Heart - focal necrosis, fragmentation of fibers
Treatment (No Specific Antidote)
- Gastric lavage with potassium permanganate (after endotracheal intubation) - oxidizes phosphine to non-toxic phosphate; repeat 2-3 times
- Activated charcoal 100 g orally mixed with sorbitol (not water) - 240 mL per 30 g to adsorb phosphine
- Antacids - reduce GI symptoms and decrease phosphine absorption
- Liquid paraffin - aids excretion of ALP and phosphine from gut
- Magnesium sulphate - reduces organ toxicity, corrects hypomagnesaemia and arrhythmias; dose: 1 g IV, repeated every 2 hours, then 1-1.5 g every 6 hours for 5-7 days as continuous IV infusion
- IV fluids for shock: 4-6 litres in first 3-6 hours (50% normal saline)
- Low-dose dopamine: 4-6 mcg/kg/min
- IV hydrocortisone: 400 mg every 4-6 hours (highly effective; reduces dopamine requirement)
- Oxygen for hypoxia
- IV sodium bicarbonate for metabolic acidosis
- Peritoneal or hemodialysis if required
- Poisoning type: Usually suicidal, occasionally accidental, rarely homicidal
Part 2: Zinc Phosphide Poisoning
- Steel-grey crystalline powder; characteristic garlicky or fishy odor
- Commonly used as grain preservative and rodenticide
- Same mechanism as ALP (liberates phosphine gas on contact with moisture)
- Slower onset than ALP due to gradual phosphine release
- Death within a few hours from pulmonary edema, or within 30 hours from cardiovascular collapse (direct myocardial toxicity)
- Treatment: same as ALP
Part 3: Anticoagulant Rodenticides
First Generation - Warfarin
- Disguised as yellow corn meal or rolled oats
- Single accidental ingestion: usually insignificant in children
- Significant coagulopathy requires large single or repeated doses
- Onset of effect: 12-48 hours after ingestion
- Biologic half-life: ~42 hours
- Single mouthful: no treatment needed
- Potentially toxic ingestion: activated charcoal
- Baseline PT/INR → repeat at 12-24 hours
- Vitamin K₁ (phytonadione) if INR >2.0: children 1-5 mg/day PO; adults 20 mg/day PO in 2-4 divided doses
Second Generation - Superwarfarins (More Important)
- Responsible for ~80% of human rodenticide exposures in the US
- Same mechanism as warfarin (inhibit Vitamin K epoxide reductase → block factors II, VII, IX, X) but far more potent and prolonged
- Brodifacoum half-life: ~120 days - a single ingestion can cause anticoagulation for weeks to months
- Coagulopathy within 24-48 hours after intentional ingestion
- Unexplained bleeding: GI bleed, hematuria, ecchymoses, intracranial hemorrhage
- Prolonged PT/INR + aPTT
- Not detected by standard warfarin assays - need specific reference lab superwarfarin assay
- Early ingestion: gastric lavage + activated charcoal
- Baseline INR → repeat at 12 and 24 hours
- If INR elevated, no active bleed: oral Vitamin K₁ (high doses may be needed for weeks to months)
- Active hemorrhage: IV Vitamin K₁ 10 mg slow infusion + four-factor prothrombin complex concentrate (4F-PCC) or 3F-PCC + fresh frozen plasma (FFP) for acute factor replacement
- Monitor INR serially after stopping Vitamin K to confirm no rebound
Quick Comparison Summary
| Feature | Aluminium Phosphide | Zinc Phosphide | Superwarfarin |
|---|---|---|---|
| Mechanism | Cytochrome oxidase inhibition (phosphine) | Same | Vitamin K epoxide reductase inhibition |
| Onset | 6-12 hrs; rapid | Slower onset | 24-48 hrs |
| Key feature | Cardiogenic shock, ARDS | Pulmonary edema, CV collapse | Prolonged bleeding/coagulopathy |
| Odor | Garlic | Garlic/fishy | None |
| Antidote | None | None | Vitamin K₁ |
| Key test | Silver nitrate paper (blackens) | Same | PT/INR, superwarfarin assay |
| Mortality | 35-100% | High | Variable (treatable) |
- The Essentials of Forensic Medicine and Toxicology, 36th ed., pp. 499-500
- Tintinalli's Emergency Medicine, pp. 1349-1350
Takayasu Arteritis what is it, its types, its causes and its mechanisms with its complications and their mechanisms, its treatment and their mechanisms all explained in detail
Takayasu Arteritis - Complete Clinical Guide
What Is It?
- "Pulseless disease" - due to absent/reduced upper limb pulses
- "Occlusive thromboarteriopathy" - due to its obliterative nature
- "Aortic arch syndrome"
- Female:male ratio 8:1 to 10:1
- Age of onset typically 10-40 years
- Most prevalent in East Asia (Japan, Korea, China, India), but global distribution
- HLA haplotype associations identified in Japanese and Mexican populations
- Incidence: approximately 1-3 per million per year
Types / Classification
| Type | Vessels Involved | Features |
|---|---|---|
| Type I (8%) | Branches from the aortic arch only | Classic "pulseless disease" - subclavian, common carotid, brachiocephalic |
| Type IIa | Ascending aorta, aortic arch and its branches | |
| Type IIb | Ascending aorta, aortic arch and branches + thoracic descending aorta | |
| Type III | Thoracic descending aorta, abdominal aorta, and/or renal arteries | |
| Type IV | Abdominal aorta and/or renal arteries only | |
| Type V (most common) | Combined features of IIb + IV - entire aorta |
- C(+) = coronary artery involvement
- P(+) = pulmonary artery involvement
The most commonly affected arteries overall are the subclavian and common carotid arteries. More than 90% of patients have stenotic/occlusive lesions; approximately 25% have aneurysms. Pulmonary arteries are involved in up to 50% of cases.
Causes and Etiology
1. Autoimmune/Immune-Mediated
- Strong association with other autoimmune diseases: rheumatoid arthritis, ankylosing spondylitis, inflammatory bowel disease
- Cell-mediated autoimmune process involving macrophages and CD4+/CD8+ T cells
- No specific autoantibody identified, but hypergammaglobulinemia is common
2. Genetic Susceptibility (HLA Association)
- High frequency of HLA haplotypes in Japanese and Mexican patients (specific alleles not uniformly replicated in North America)
- HLA-B52 is the most consistently associated allele
- Genetic susceptibility varies by geography, which may partially explain geographic differences in prevalence
3. Infectious Trigger (Proposed)
- Mycobacterium tuberculosis has been long proposed as a trigger - there is geographic overlap between TA and TB-endemic regions
- Molecular mimicry: mycobacterial heat-shock protein (HSP65) shares antigenic epitopes with aortic smooth muscle cells
- However, this causal link remains unproven
4. Estrogen/Sex Hormones
- The overwhelming female predominance and peak in reproductive age suggests sex hormone modulation of immune responses, though mechanisms are not fully defined
Pathogenesis / Mechanism of Disease
Step 1: Antigen Presentation and Dendritic Cell Activation
- An unknown antigen (possibly from vascular wall or exogenous trigger) is presented by dendritic cells in the adventitia of large arteries
- Dendritic cells in the vasa vasorum (small vessels that supply the arterial wall) serve as the gateway for immune cells entering the vessel wall
- Activated dendritic cells recruit and activate CD4+ and CD8+ T lymphocytes
Step 2: T-Cell Mediated Inflammatory Cascade
- Th1 pathway: produces IFN-γ and IL-12, activating macrophages - this pathway is relatively corticosteroid-resistant
- Th17 pathway: produces IL-17, promoting further inflammatory recruitment - this pathway is corticosteroid-sensitive
- CD8+ cytotoxic T cells and NK cells directly kill vascular smooth muscle cells via perforin/granzyme release
- B lymphocytes and plasma cells also accumulate
Step 3: Granuloma Formation and Giant Cells
- Macrophages fuse to form multinucleated giant cells - the histopathologic hallmark
- These collect particularly around the vasa vasorum in the adventitia and outer media
- Granulomatous inflammation causes patchy medial necrosis and erosion of elastic and smooth muscle tissue
Step 4: Structural Consequences
- Destruction of medial smooth muscle → replaced by transmural collagenous fibrosis
- Concurrent intimal hyperplasia (growth factor-driven mesenchymal/myofibroblast proliferation)
- Progressive luminal narrowing and obliteration → end-organ ischemia
- Matrix metalloproteinase (MMP) synthesis (by inflammatory cells) degrades the extracellular matrix and elastic tissue
- Loss of structural support in media → aneurysmal dilation
Role of IL-6
- IL-6 is a key cytokine in TA, produced by macrophages and vascular wall cells
- Drives acute-phase response (CRP, ESR elevation), fever, and promotes Th17 differentiation
- This is the mechanistic rationale for tocilizumab (anti-IL-6R) therapy
Histopathology / Morphology
- Classic "tree-bark" surface of the aorta
- Irregular thickening of the vessel wall
- Patchy involvement with "skip lesions" is the most common pattern
- Great vessel lumens can be markedly narrowed or obliterated
- Early/active: Adventitial mononuclear infiltrates with perivascular cuffing of the vasa vasorum; lymphocytes, histiocytes, macrophages, plasma cells; PMNs and multinucleated giant cells around vasa vasorum; patchy medial necrosis
- Progressive: Granulomatous inflammation replete with giant cells throughout all three layers (indistinguishable from giant cell arteritis histologically)
- Late/healed: Collagenous scarring with admixed chronic inflammatory infiltrates in all three layers; intimal hyperplasia
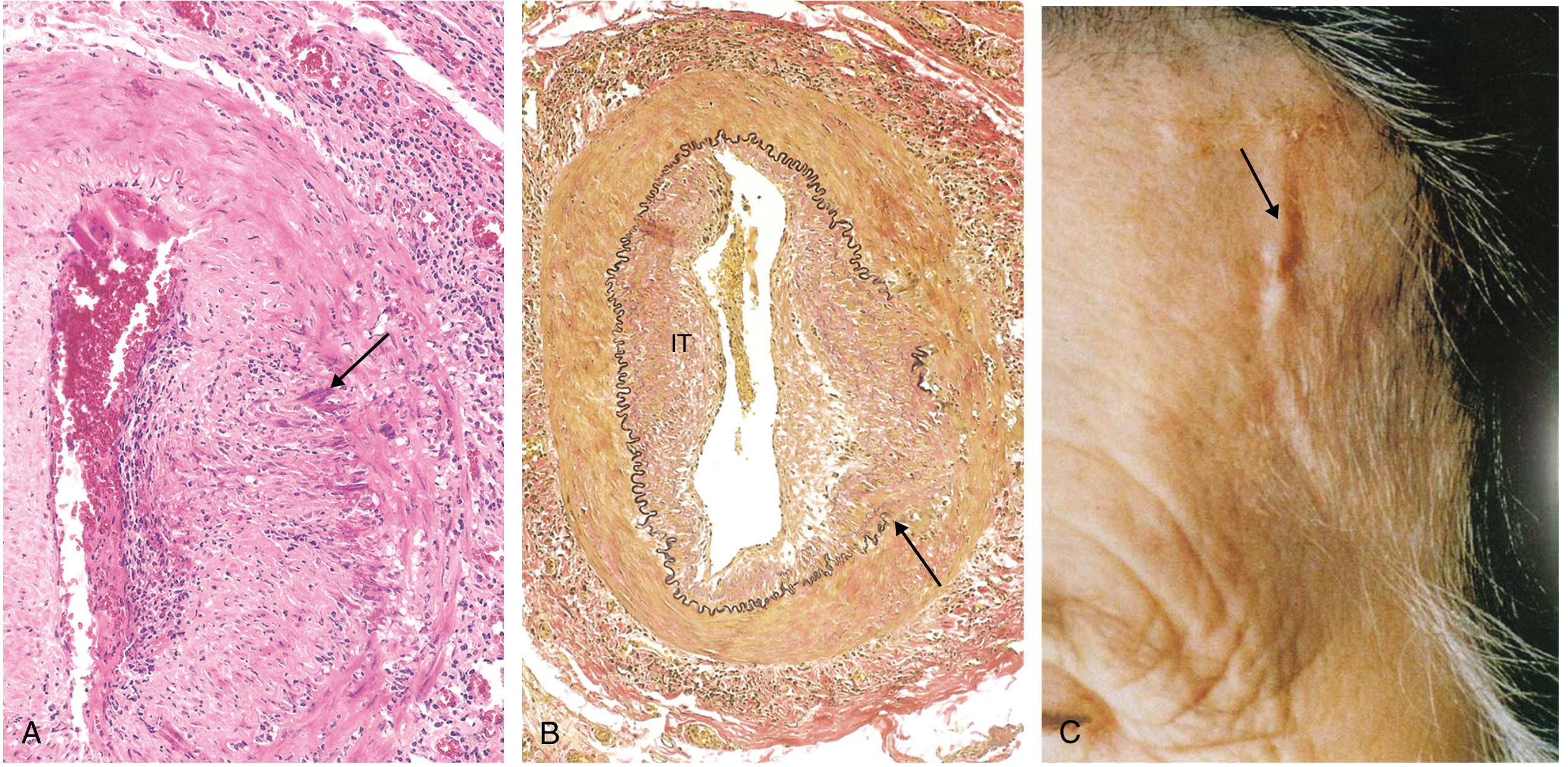
Note: The histology of TA and giant cell arteritis (GCA) is essentially indistinguishable. The key differentiator is age: TA is diagnosed in patients under 50; GCA in those over 50.
Clinical Phases and Presentation
Phase 1: "Pre-pulseless" / Systemic Phase (Early)
- Fever, night sweats, malaise, profound tiredness, lethargy
- Anorexia, weight loss, arthralgias
- Skin rash (erythema nodosum-like)
- Lab: ESR and CRP elevated; normochromic normocytic anemia; thrombocytosis
Phase 2: "Pulseless" / Vascular Phase (Late)
| Artery Involved | Clinical Feature |
|---|---|
| Subclavian / brachial | Absent/weak pulses, asymmetric BP (>10 mmHg difference), upper limb claudication |
| Common carotid | Carotidynia (in 25%), bruits |
| Vertebral / carotid | Dizziness, syncope, vertigo, hemiparesis, stroke |
| Ophthalmic / retinal | Visual blurring, diplopia, amaurosis fugax, blindness, retinal haemorrhage, optic atrophy |
| Renal arteries | Systemic hypertension (in ~50%), CKD |
| Ascending aorta/root | Aortic regurgitation |
| Coronary ostia | Angina, MI (often silent) |
| Pulmonary arteries | Pulmonary hypertension |
| Abdominal aorta / mesenteric | Intestinal angina, GI bleeding |
| Lower limb arteries | Leg claudication, rest pain |
Complications and Their Mechanisms
1. Stroke / Cerebrovascular Events
2. Aortic Regurgitation (AR)
3. Systemic Hypertension
4. Pulmonary Hypertension
5. Myocardial Infarction / Coronary Arteritis
6. Heart Failure
7. Aortic Aneurysm and Rupture
8. Renal Failure / CKD
9. Blindness / Visual Loss
10. Anastomotic Aneurysms (Post-surgical)
Diagnosis
ACR 1990 Diagnostic Criteria (≥3 of 6 = >90% sensitivity and specificity)
| Criterion | Definition |
|---|---|
| Age at onset | <40 years |
| Claudication | Upper or lower extremity fatigue with exercise |
| Diminished brachial pulse | Unilateral or bilateral |
| Asymmetric brachial BP | >10 mmHg difference between arms |
| Bruit | Audible over aorta or subclavian artery |
| Angiographic abnormalities | Stenosis/occlusion of aorta or major branches (not from atherosclerosis/FMD) |
Investigations
- Lab: ESR, CRP (raised in 75%); normochromic normocytic anaemia; thrombocytosis; hypergammaglobulinemia; hypoalbuminaemia. No specific autoantibody.
- Imaging (gold standard now non-invasive):
- MRA / CTA: preferred - shows vessel wall enhancement, edema, thickening, stenoses and aneurysms; MRA preferred for serial monitoring (no radiation)
- 18F-FDG PET-CT: detects active metabolic arteritis before stenosis develops; useful for pre-stenotic disease
- Colour duplex ultrasound: excellent for common carotid and proximal subclavian arteries; shows "halo sign" - hypoechoic circumferential wall thickening
- Conventional angiography: historical gold standard; still used when endovascular intervention is planned
- Coronary CTA: best for detecting coronary ostial involvement (neither MRA nor PET reliably identifies this)
Treatment and Mechanisms
1. Glucocorticoids (First Line)
- Suppress the Th17/IL-17 pathway rapidly and effectively
- Reduce dendritic cell activation and cytokine production (TNF-α, IL-1, IL-6)
- Decrease vascular wall inflammation, edema, and acute-phase response
- Limitation: Th1/IFN-γ pathway is relatively corticosteroid-resistant, explaining the high relapse rate (~50%) on tapering
2. Methotrexate (Most Widely Used Steroid-Sparing Agent)
- Inhibits dihydrofolate reductase (DHFR) → depletes reduced folates → impairs DNA synthesis and cell proliferation of rapidly dividing immune cells
- Reduces T-cell and B-cell activation and cytokine production
- Allows prednisolone dose reduction
- Small open-label studies support use; most widely prescribed alongside azathioprine and MMF
3. Azathioprine / Mycophenolate Mofetil (MMF) (Steroid-Sparing)
- Prodrug converted to 6-mercaptopurine → inhibits purine synthesis → suppresses T and B cell proliferation
- Inhibits inosine monophosphate dehydrogenase (IMPDH) → selectively blocks de novo guanosine nucleotide synthesis in lymphocytes (other cells have a salvage pathway) → suppresses T and B cell proliferation
4. Cyclophosphamide (For Refractory or Life-Threatening Disease)
- Alkylating agent - cross-links DNA strands via its nitrogen mustard moiety → inhibits DNA replication → kills rapidly dividing cells, especially lymphocytes
- Profoundly immunosuppressive, effective in refractory cases
- Given as IV pulsed cyclophosphamide typically for 6 months, then switched to azathioprine for maintenance
5. Anti-TNF-α Agents (Infliximab, Etanercept) - For Refractory TA
- Bind and neutralize TNF-α, a key macrophage-derived pro-inflammatory cytokine
- TNF-α drives granuloma formation, macrophage activation, and vascular wall inflammation
- Published case review: complete remission 37%, partial remission 53.5%, no response 9.5% with TNF-α antagonists in TA
6. Tocilizumab (Anti-IL-6 Receptor) - For Refractory TA
- Monoclonal antibody that blocks the IL-6 receptor (IL-6R)
- IL-6 drives: acute-phase response (CRP, fibrinogen), Th17 differentiation, macrophage activation, vascular smooth muscle cell proliferation
- Blocking IL-6R suppresses all these downstream effects
- Effective in refractory TA; an initial placebo-controlled trial showed beneficial effect
- Important caveat: Tocilizumab also suppresses CRP synthesis independent of disease activity, so CRP can normalize even in active disease - angiographic monitoring (MRI) is mandatory
7. Aspirin and Antiplatelet Therapy
8. Antihypertensive Therapy
9. Surgical and Endovascular Intervention
- Aneurysmal enlargement with rupture risk
- Severe aortic regurgitation
- Stenoses causing: severe coronary or cerebrovascular disease, uncontrolled renovascular hypertension, critical limb ischemia
- Open surgical bypass: Best long-term patency. Must anastomose to non-diseased vessel segments - anastomosis to diseased wall risks pseudoaneurysm (12% at 20 years). Avoid when possible until active inflammation is controlled.
- Percutaneous transluminal angioplasty (PTA) ± stenting: Higher restenosis rates than open surgery; useful as temporizing strategy
- Aortic valve + root replacement: For severe AR due to aortic root dilation
Prognosis
- 15-year survival: >95% overall in Japan and US (96.5% in Japan)
- Falls to 67% in patients with serious complications or progressive disease course
- 74% report compromised daily activities; 23% unable to work
- Scar tissue and vascular obstruction persist after disease control (fibrosis is irreversible) - this is why structural complications still require surgery even in remission
Takayasu Arteritis what is it, its types, its causes and its mechanisms with its complications and their mechanisms, its treatment and their mechanisms all explained in detail
Takayasu Arteritis - Complete Clinical Guide
What Is It?
- "Pulseless disease" - absent/reduced upper limb pulses
- "Occlusive thromboarteriopathy" - obliterative nature
- "Aortic arch syndrome"
- Female:male ratio 8:1 to 10:1
- Age of onset typically 10-40 years
- Most prevalent in East Asia but global distribution
- HLA haplotype associations in Japanese and Mexican populations
Types / Classification (Numano Angiographic Classification)
| Type | Vessels Involved |
|---|---|
| Type I (8%) | Branches from the aortic arch only (subclavian, carotid, brachiocephalic) |
| Type IIa | Ascending aorta, aortic arch and its branches |
| Type IIb | Ascending aorta, aortic arch + branches + thoracic descending aorta |
| Type III | Thoracic descending aorta, abdominal aorta, and/or renal arteries |
| Type IV | Abdominal aorta and/or renal arteries only |
| Type V (most common) | Combined IIb + IV - entire aorta involved |
Causes and Etiology
1. Autoimmune/Immune-Mediated
- Strong association with rheumatoid arthritis, ankylosing spondylitis, inflammatory bowel disease
- Cell-mediated process driven by macrophages and CD4+/CD8+ T cells
- No specific autoantibody identified
2. Genetic Susceptibility (HLA)
- HLA-B52 is the most consistently associated allele
- HLA haplotype links found in Japanese and Mexican patients
- Genetic variation explains geographic differences in prevalence
3. Infectious Trigger (Proposed)
- Mycobacterium tuberculosis proposed as a trigger via molecular mimicry - mycobacterial heat-shock protein (HSP65) shares antigenic epitopes with aortic smooth muscle cells
- Geographic overlap between TA and TB-endemic regions
- Causal link remains unproven
4. Sex Hormones
- Overwhelming female predominance and peak in reproductive years suggests sex hormone modulation of immune responses
Pathogenesis - Step-by-Step Mechanism
Step 1: Antigen Presentation via Vasa Vasorum
- An unknown antigen triggers dendritic cells in the adventitia/vasa vasorum (the small vessels supplying the arterial wall)
- Activated dendritic cells present antigen to CD4+ and CD8+ T lymphocytes
Step 2: Two T-Cell Pathways Activated
| Pathway | Cytokines | Effect | Steroid Sensitivity |
|---|---|---|---|
| Th1 | IL-12, IFN-γ | Macrophage activation, granuloma formation | Resistant |
| Th17 | IL-17 | Inflammatory cell recruitment | Sensitive |
- CD8+ cytotoxic T cells and NK cells directly kill vascular smooth muscle cells via perforin/granzyme
- B cells and plasma cells also accumulate
- IL-6 is a key cytokine driving the acute-phase response and Th17 differentiation
Step 3: Granuloma Formation
- Macrophages fuse → multinucleated giant cells (histopathologic hallmark)
- Giant cells collect around vasa vasorum in adventitia and outer media
- Granulomatous inflammation causes patchy medial necrosis and elastic tissue erosion
Step 4: Structural Consequences (Two Opposing Outcomes)
- Media destruction → replaced by transmural collagenous fibrosis
- Concurrent intimal hyperplasia (growth factor-driven myofibroblast proliferation)
- Progressive luminal narrowing → end-organ ischemia
- Matrix metalloproteinase (MMP) synthesis by inflammatory cells degrades extracellular matrix and elastic tissue
- Loss of medial structural support → aneurysmal dilation
Histopathology
- Early/active: Adventitial mononuclear infiltrates; lymphocytes, histiocytes, macrophages, plasma cells; PMNs and multinucleated giant cells around vasa vasorum; patchy medial necrosis
- Progressive: Full granulomatous inflammation with giant cells in all three wall layers
- Late/healed: Collagenous scarring throughout all layers; intimal hyperplasia; lumen near-obliterated
The histology of TA is indistinguishable from giant cell arteritis (GCA). Age is the key differentiator: TA = under 50 years; GCA = over 50 years.
Clinical Phases and Presentation
Phase 1: "Pre-pulseless" / Systemic Phase
- Fever, night sweats, malaise, fatigue, weight loss, arthralgia
- Skin rash (erythema nodosum-like)
- Labs: Elevated ESR and CRP (75%); normochromic normocytic anaemia; thrombocytosis
Phase 2: "Pulseless" / Vascular Phase
| Artery | Clinical Feature |
|---|---|
| Subclavian/brachial | Absent/weak pulses, asymmetric BP >10 mmHg, upper limb claudication |
| Common carotid | Carotidynia (25%), bruits |
| Vertebral/carotid | Dizziness, syncope, vertigo, hemiparesis, stroke |
| Ophthalmic/retinal | Visual blurring, diplopia, amaurosis fugax, blindness, optic atrophy |
| Renal arteries | Systemic hypertension (~50%), CKD |
| Ascending aorta/root | Aortic regurgitation |
| Coronary ostia | Angina, MI (often silent) |
| Pulmonary arteries | Pulmonary hypertension |
| Abdominal aorta | Intestinal angina, GI bleeding, leg claudication |
Complications and Their Mechanisms
1. Stroke
2. Aortic Regurgitation (18% of patients; 15% need valve replacement)
3. Systemic Hypertension
4. Pulmonary Hypertension
5. Myocardial Infarction / Coronary Arteritis
6. Heart Failure
7. Aortic Aneurysm and Rupture
8. Renal Failure / CKD
9. Blindness
10. Anastomotic Aneurysms (Post-surgical)
Diagnosis
ACR 1990 Criteria (≥ 3 of 6 required = >90% sensitivity/specificity)
| Criterion | Definition |
|---|---|
| Age at onset | < 40 years |
| Claudication | Upper or lower limb fatigue with exercise |
| Diminished brachial pulse | Unilateral or bilateral |
| Asymmetric brachial BP | > 10 mmHg difference |
| Bruit | Over aorta or subclavian artery |
| Angiographic abnormalities | Stenosis/occlusion not from atherosclerosis/FMD |
Investigations
- Labs: ESR, CRP (elevated 75%); normochromic normocytic anaemia; thrombocytosis; no specific autoantibody
- MRA / CTA: Preferred imaging - shows wall thickening, enhancement, edema, stenoses, aneurysms. MRA preferred for serial monitoring (no radiation)
- 18F-FDG PET-CT: Detects active arteritis metabolically before stenosis develops; useful for pre-stenotic disease
- Colour duplex ultrasound: "Halo sign" - hypoechoic circumferential wall thickening of carotid/subclavian arteries
- Coronary CTA: Best for coronary ostial involvement (neither MRA nor PET is reliable here)
- Conventional angiography: Historical gold standard; still used when endovascular intervention is planned
Treatment and Mechanisms (Detailed)
1. Glucocorticoids (First Line)
- Suppress Th17/IL-17 pathway - rapidly and effectively
- Reduce dendritic cell activation and production of TNF-α, IL-1, IL-6
- Decrease vascular wall edema and acute-phase response
- Key limitation: The Th1/IFN-γ pathway is corticosteroid-resistant → explains ~50% relapse rate on tapering
- 86% of patients experience glucocorticoid-related adverse events at 10-year follow-up → mandatory steroid-sparing strategy
2. Methotrexate (Most Widely Used Steroid-Sparing Agent)
- Inhibits dihydrofolate reductase (DHFR) → depletes reduced folate → impairs DNA/RNA synthesis in rapidly dividing immune cells
- Reduces T-cell and B-cell proliferation and cytokine production (including IL-6, IL-1)
- Allows meaningful prednisolone dose reduction
- Most widely prescribed steroid-sparing agent in TA
3. Azathioprine (Steroid-Sparing)
- Prodrug converted to 6-mercaptopurine (6-MP) → inhibits purine synthesis (specifically HGPRT pathway) → blocks DNA synthesis in lymphocytes
- Suppresses both T and B cell proliferation
4. Mycophenolate Mofetil / MMF (Steroid-Sparing)
- Inhibits inosine monophosphate dehydrogenase (IMPDH) → selectively blocks de novo guanosine nucleotide synthesis in lymphocytes (other cells use salvage pathway)
- Highly selective lymphocyte suppression with relatively less systemic toxicity
5. Cyclophosphamide (Refractory / Life-Threatening Disease)
- Alkylating agent - nitrogen mustard derivative cross-links DNA strands → prevents DNA replication → kills rapidly dividing cells, especially lymphocytes
- Given as IV pulsed cyclophosphamide for ~6 months then switched to azathioprine for maintenance
- Most potent immunosuppressant available for TA
6. Anti-TNF-α Agents (Infliximab, Etanercept) - Refractory TA
- Bind and neutralize TNF-α - a key macrophage/T-cell pro-inflammatory cytokine
- TNF-α drives granuloma formation, macrophage activation, NF-κB signaling, and vascular wall inflammation
- Review of all published TA cases: complete remission 37%, partial remission 53.5%, no response 9.5%
7. Tocilizumab (Anti-IL-6 Receptor) - Refractory TA
- Monoclonal antibody blocking the IL-6 receptor (IL-6R)
- IL-6 drives: CRP/acute-phase response, Th17 differentiation, macrophage activation, smooth muscle cell proliferation
- Blocking IL-6R suppresses all these downstream effects
- A placebo-controlled trial in TA showed beneficial effect; GIACTA trial strongly supports use in GCA
8. Aspirin / Antiplatelet Therapy
9. Antihypertensives (for Renovascular Hypertension)
10. Surgical / Endovascular Intervention
- Aneurysmal enlargement with rupture risk
- Severe aortic regurgitation
- Stenoses causing: severe coronary/cerebrovascular disease, uncontrolled renovascular hypertension, critical limb ischemia
| Intervention | Mechanism | Key Point |
|---|---|---|
| Open surgical bypass | Creates new flow conduit bypassing occluded segment | Best long-term patency; anastomose only to non-diseased vessel segments (diseased wall → pseudoaneurysm) |
| PTA ± stenting | Balloon dilation mechanically expands stenotic segment | Higher restenosis rates; useful temporizing measure |
| Aortic valve + root replacement | Removes diseased, incompetent valve and dilated root | For severe AR; graft replaces aortic root |
Prognosis
- 15-year survival: >95-96% in Japan and US; 87% in Korea at 10 years
- Falls to 67% with serious complications or progressive disease
- 74% report compromised daily activities; 23% are unable to work
- Structural complications (stenoses, fibrosis) persist after disease control - fibrosis is irreversible, hence why structural interventions are often still needed even in immunologic remission
Albumin in critically ill patients journals
albumin infusion critically ill patients outcomes
albumin resuscitation sepsis ICU mortality
albumin critically ill ICU mortality systematic review
albumin supplementation hypoalbuminemia ICU randomized trial
ATTIRE trial albumin critically ill
albumin versus saline fluid resuscitation ICU SAFE ALBIOS
human albumin solution critically ill review evidence guidelines
PMID: 40600034
PMID: 40020556
PMID: 39879912
PMID: 40885608
albumin sepsis liver failure hepatic critically ill
PMID: 34036519
SAFE ALBIOS ATTIRE TACTICS albumin critically ill trials summary 2024 2025
albumin critically ill ICU 2024 2025 guidelines CHEST ESICM current evidence
Albumin in Critically Ill Patients - Evidence from Key Journals
Why Albumin? Physiological Rationale
- Oncotic effect: draws fluid back into the intravascular compartment
- Drug and molecule binding: transports fatty acids, hormones, drugs
- Antioxidant/free-radical scavenging: thiol groups on albumin neutralize reactive oxygen species
- Nitric oxide (NO) modulation: reduces vasodilation in sepsis
- Acid-base buffering: the negative charge of albumin provides anionic buffer capacity
- Anti-inflammatory effects: limits endothelial glycocalyx shedding
Landmark Trials - Chronological Summary
1. SAFE Study (2004) - NEJM - The Foundation
- No significant difference in 28-day mortality (20.9% albumin vs. 21.1% saline; RR 0.99)
- Albumin was safe overall
- Pre-specified subgroup (sepsis): trend toward lower mortality with albumin (30.7% vs. 35.3%; RR 0.87; p=0.09) - not statistically significant but generated the hypothesis
- Critical exception - TBI: albumin-treated TBI patients had significantly higher mortality (33.2% vs. 20.4%; RR 1.63; p=0.003) - albumin is CONTRAINDICATED in TBI
2. ALBIOS Trial (2014) - NEJM - The Targeted Replacement Trial
| Outcome | Albumin group | Crystalloid group | p value |
|---|---|---|---|
| Death at 28 days (primary) | 31.8% (285/895) | 32.0% (288/900) | RR 1.00; p=0.94 |
| Death at 90 days | 41.1% | 43.6% | RR 0.94; p=0.29 |
| SOFA score change | Improved | Less improved | p=0.03 |
| MAP at 6 hours | 79 vs 77 mmHg | - | p<0.001 |
| Net fluid balance Day 7 | +350 mL | +1,220 mL | Less positive |
- Achieved target: albumin ≥30 g/L (day 1: 28.6 vs 24.0 g/L; p<0.001)
- Pre-specified subgroup (septic shock): 90-day mortality 43.6% vs. 49.9% (p=0.03) - significant survival benefit in the sickest patients
- Overall conclusion: No benefit in all-comers with severe sepsis. Signal of benefit in septic shock specifically.
3. FRISC Study (2021) - Hepatology International - Cirrhosis + Sepsis
- Reversal of hypotension (MAP ≥65 mmHg) at 1 hour: 25.3% (albumin) vs. 11.7% (saline) (OR 1.9; p=0.03)
- Reversal at 3 hours: 11.7% vs. 3.2% (OR 3.9; p=0.008)
- Sustained lactate clearance: better in albumin group (p<0.001)
- 1-week survival: 43.5% vs. 38.3% (p=0.03)
4. ATTIRE Trial (2021) - NEJM - Cirrhosis Inpatients
- No significant reduction in infection, renal impairment, or mortality (primary composite endpoint: 29% vs. 30%; p=0.72)
- Increased serious adverse events in albumin group, including pulmonary edema and fluid overload
- Post-hoc AASLD 2023 data: high doses of albumin increased mortality and complications in terlipressin-treated cirrhotic patients
5. Fluid Resuscitation NMA (2025) - Front Med - Network Meta-Analysis
- Balanced crystalloids: lowest all-cause mortality (SUCRA 83.1%), shortest ICU and hospital stay → preferred first-line fluid
- Hyper-oncotic albumin (20%): lowest renal replacement therapy events (SUCRA 94.1%)
- H-HES (high-MW hydroxyethyl starch): worst outcomes - increased mortality, AKI, RRT → strongly advise against
- Albumin not ranked as best overall but notable RRT advantage
6. 20% Albumin in Sepsis Microcirculation RCT (2025) - J Crit Care
- 20% albumin produced significant improvements in microvascular density and activity at 15 min and 60 min (p<0.005)
- Crystalloid group: no significant microcirculatory changes
- No difference in overall fluid balance, vasopressor days, ICU stay, or mortality
7. Albumin in Severe Acute Pancreatitis (2025) - Pancreatology
- No difference in SIRS remission or 60-day mortality
- Lower incidence of sepsis in albumin group (10% vs. 36.7%; p=0.01)
8. Expert Consensus Review (2025) - Heart & Lung
- Crystalloids remain preferred first-line fluid for sepsis - safe, cost-effective, available
- Albumin conditionally recommended in specific scenarios:
- Severe hypoalbuminemia
- High vasopressor requirements
- Volume-sensitive conditions
- Individualized management based on patient-specific factors and dynamic monitoring is emphasized
- Guidelines advise against routine albumin use
9. ICTM Guidelines (2024) - CHEST Journal
| Indication | Recommendation |
|---|---|
| Critically ill adults (non-burn, non-ARDS) - first-line resuscitation | NOT suggested |
| Critically ill adults - targeted albumin replacement | NOT suggested |
| Patients with cirrhosis - large volume paracentesis (>5L) | Conditional YES (low certainty) |
| Cirrhosis + spontaneous bacterial peritonitis (SBP) | Conditional YES (low certainty) |
| Decompensated cirrhosis + hypoalbuminemia | NOT suggested |
| Cirrhosis + extraperitoneal infections | NOT suggested |
| Traumatic brain injury | NOT suggested (HARM signal) |
| Cardiac surgery (bypass priming/volume) | NOT suggested |
| Pediatric/neonatal critical care | NOT suggested in most scenarios |
Disease-Specific Summary
Sepsis / Septic Shock
| Context | Evidence | Bottom Line |
|---|---|---|
| General severe sepsis | ALBIOS: no 28-day mortality benefit | Not routine |
| Septic shock specifically | ALBIOS subgroup: 90-day benefit (p=0.03) | May consider 20% albumin if serum albumin <26 g/L |
| Microcirculation | 2025 RCT: significant improvement with 20% albumin | Mechanistic rationale for sickest patients |
| Network MA (2025) | Balanced crystalloids best overall; albumin not first-line | Use balanced crystalloids first |
Cirrhosis
| Indication | Evidence | Bottom Line |
|---|---|---|
| SBP | Sort et al. 1999: reduces renal impairment + mortality | Recommended (1.5 g/kg Day 1, 1 g/kg Day 3) |
| Large-volume paracentesis (>5L) | Reduces paracentesis-induced circulatory dysfunction | Recommended (6-8 g/L drained) |
| Sepsis-induced hypotension in cirrhosis | FRISC 2021: superior to saline for reversal | Consider over saline |
| Decompensated cirrhosis - routine supplementation | ATTIRE 2021: no benefit, increased pulmonary edema | Avoid |
| Terlipressin + cirrhosis | ATTIRE 2023 post-hoc: increased mortality with high-dose albumin | Caution/Avoid |
Traumatic Brain Injury
- SAFE subgroup: albumin associated with higher mortality (RR 1.63; p=0.003)
- Absolute contraindication in TBI resuscitation
Acute Pancreatitis
- 2025 RCT (n=60): no SIRS or mortality benefit; reduced secondary sepsis
- Insufficient evidence; not recommended routinely
Current Position (2025-2026)
"Few evidence-based indications support the routine use of albumin in clinical practice to improve patient outcomes."
- Cirrhosis + SBP (definitive indication, well-established)
- Cirrhosis + large volume paracentesis >5L
- Refractory septic shock with documented severe hypoalbuminemia (<26 g/L) and high vasopressor requirement
- When 20% albumin is chosen over 5% (smaller volumes, microcirculatory benefit data)
- General ICU fluid resuscitation
- Decompensated cirrhosis with hypoalbuminemia (ATTIRE)
- Traumatic brain injury (SAFE subgroup)
- Cardiac surgery
- Routine supplementation targeting a specific serum level
Key Papers for Reference
| Trial/Study | PMID | Year | Key Finding |
|---|---|---|---|
| SAFE Study | 15163774 | 2004 | Albumin = saline in general ICU; harm in TBI |
| ALBIOS | 24635772 | 2014 | No 28-day benefit; possible benefit in septic shock |
| FRISC | 34036519 | 2021 | Albumin superior to saline in cirrhosis + sepsis |
| ATTIRE | 33626252 | 2021 | No benefit + harm in decompensated cirrhosis |
| ICTM Guideline (CHEST) | 38447639 | 2024 | 14 recommendations; only 2 conditional for albumin |
| NMA Sepsis fluids | 40600034 | 2025 | Balanced crystalloids best; hyper-oncotic albumin low RRT |
| Microcirculation RCT | 40020556 | 2025 | 20% albumin improves microcirculation in sepsis |
| Expert consensus | 39879912 | 2025 | Crystalloids first; albumin selective/individualized |
| Pancreatitis RCT | 40885608 | 2025 | No SIRS/mortality benefit; lower secondary sepsis |