MI pathogenesis explain with pictures
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Loading Image
Loading Image
Loading Image
Reading File
Loading Image
Reading File
Loading Image
Reading File
Loading Image
I now have all the key images and textbook content needed. Here is a comprehensive explanation:
Myocardial Infarction (MI) - Pathogenesis
Definition
MI ("heart attack") is necrosis of cardiac muscle resulting from prolonged ischemia. It is the death of myocytes when blood supply is cut off for long enough that irreversible injury occurs. The 2018 joint task force defines it as "acute myocardial injury detected by abnormal cardiac biomarkers in the setting of acute myocardial ischemia."
- Robbins & Kumar Basic Pathology, Robbins Cotran Pathologic Basis of Disease
Step 1 - Atherosclerotic Plaque Formation
The fundamental prerequisite for most MIs is coronary atherosclerosis. Lipid-rich plaques develop in the coronary artery walls over years. The coronary involved before the thrombus was often <70% stenosed - meaning it was not severely flow-limiting beforehand; the acute event is what causes MI.
Microscopic: Coronary artery cross-sections (normal vs. severely diseased)
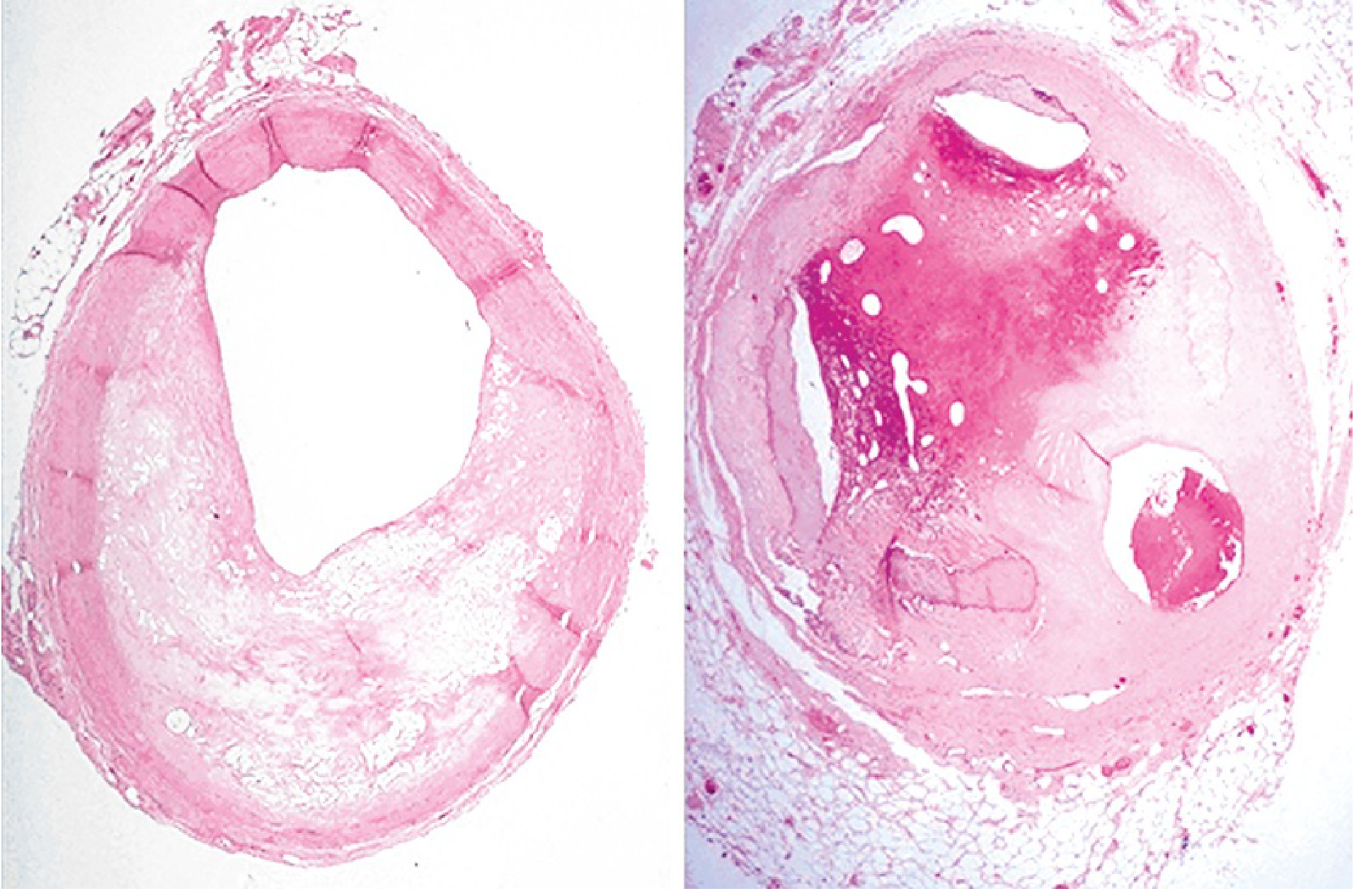
eFig. 9.1 - Left: coronary narrowed ~65%. Right: almost fully occluded by previous organized thrombus. (Robbins & Kumar Basic Pathology)
Step 2 - Plaque Disruption (the Trigger)
The classic sequence:
-
Plaque erosion or rupture - caused by endothelial injury, intraplaque hemorrhage, or mechanical shear forces. This exposes subendothelial collagen and the necrotic lipid-rich core to the bloodstream.
-
Platelet adhesion and activation - platelets stick to exposed collagen, aggregate, and release thromboxane A2, ADP, and serotonin, causing further platelet recruitment and vasospasm.
-
Coagulation cascade activation - tissue factor exposure drives thrombin generation, fibrin deposition, and rapid thrombus growth.
-
Complete luminal occlusion - within minutes the thrombus can occlude the coronary artery entirely.
Angiography within 4 hours of MI onset demonstrates coronary thrombosis in nearly 90% of cases. After 12-24 hours this falls to ~60%, as some thrombi lyse spontaneously.
Microscopic: Coronary thrombosis on a ruptured plaque
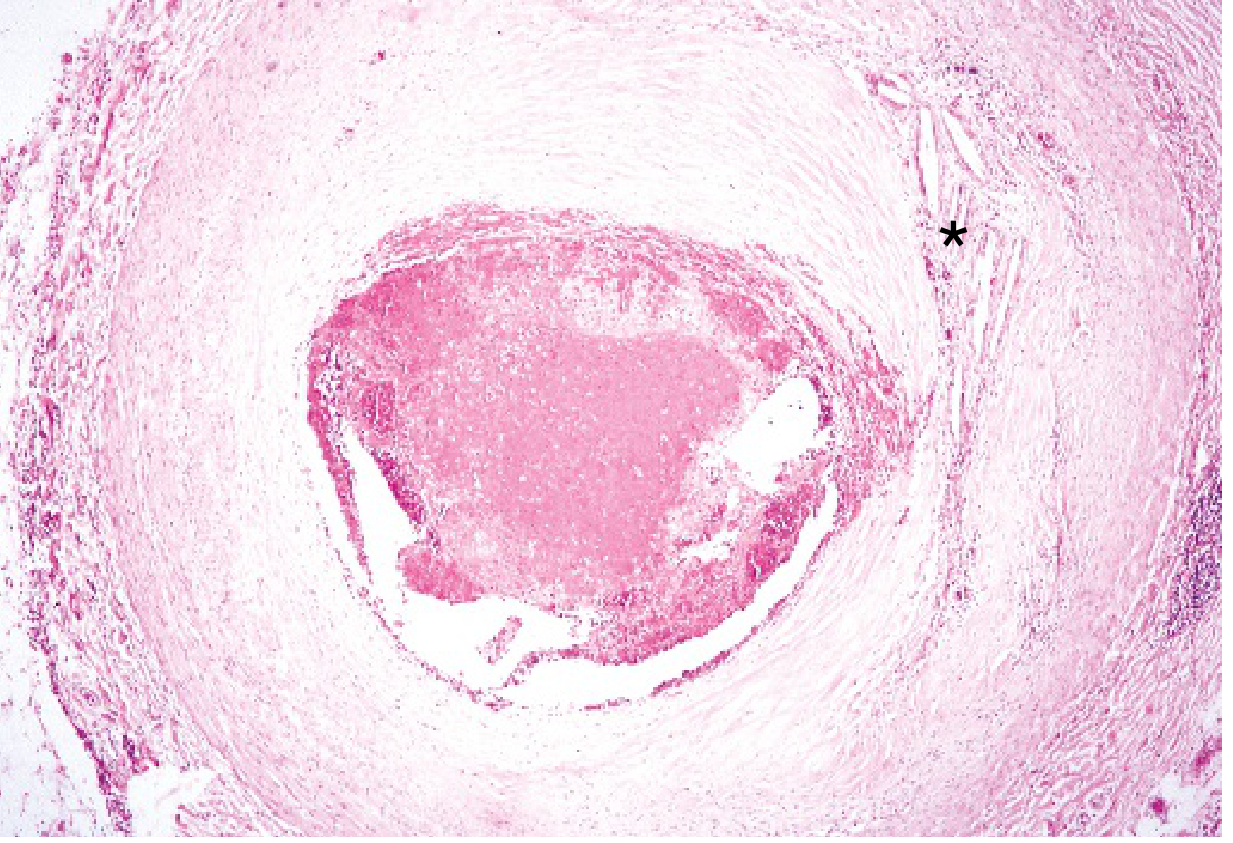
eFig. 9.2 - A markedly narrowed coronary artery nearly occluded by a fresh thrombus. Cholesterol clefts () visible in the fibrointimal plaque. (Robbins & Kumar Basic Pathology)*
Step 3 - Myocardial Response to Ischemia
Once the coronary is occluded:
| Time | Event |
|---|---|
| Seconds | Aerobic metabolism stops; ATP drops; toxic metabolites (lactate) accumulate |
| Minutes | Loss of contractility (reversible) |
| 20-40 min | Irreversible cell injury begins - coagulative necrosis of myocytes |
| Earliest sign | Sarcolemmal disruption → cardiac enzymes (troponin, CK-MB) leak into blood |
- Robbins & Kumar Basic Pathology
The sequence begins in the subendocardium (most vulnerable - furthest from epicardial vessels, highest intramural pressure) and spreads outward as a wavefront over 3-6 hours until the full wall thickness is involved (transmural infarct) if not treated.
Step 4 - Progression of Necrosis (Wavefront Phenomenon)
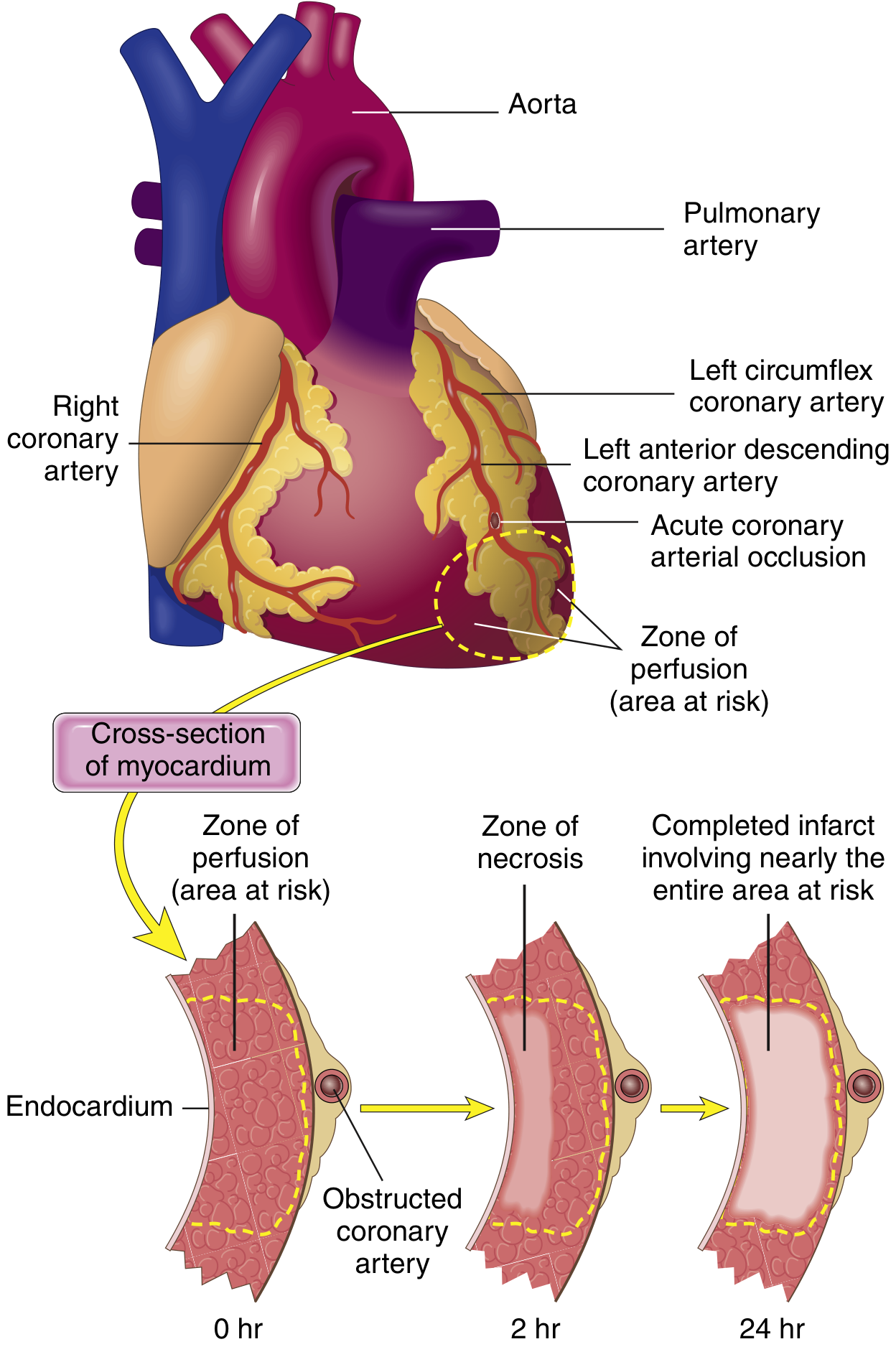
FIG. 9.8 - Necrosis begins subendocardially (2 hrs) and expands to full wall thickness (24 hrs) without intervention. A thin subendocardial rim survives via oxygen diffusion from ventricular cavity. (Robbins & Kumar Basic Pathology)
Step 5 - Patterns of Infarction by Vessel
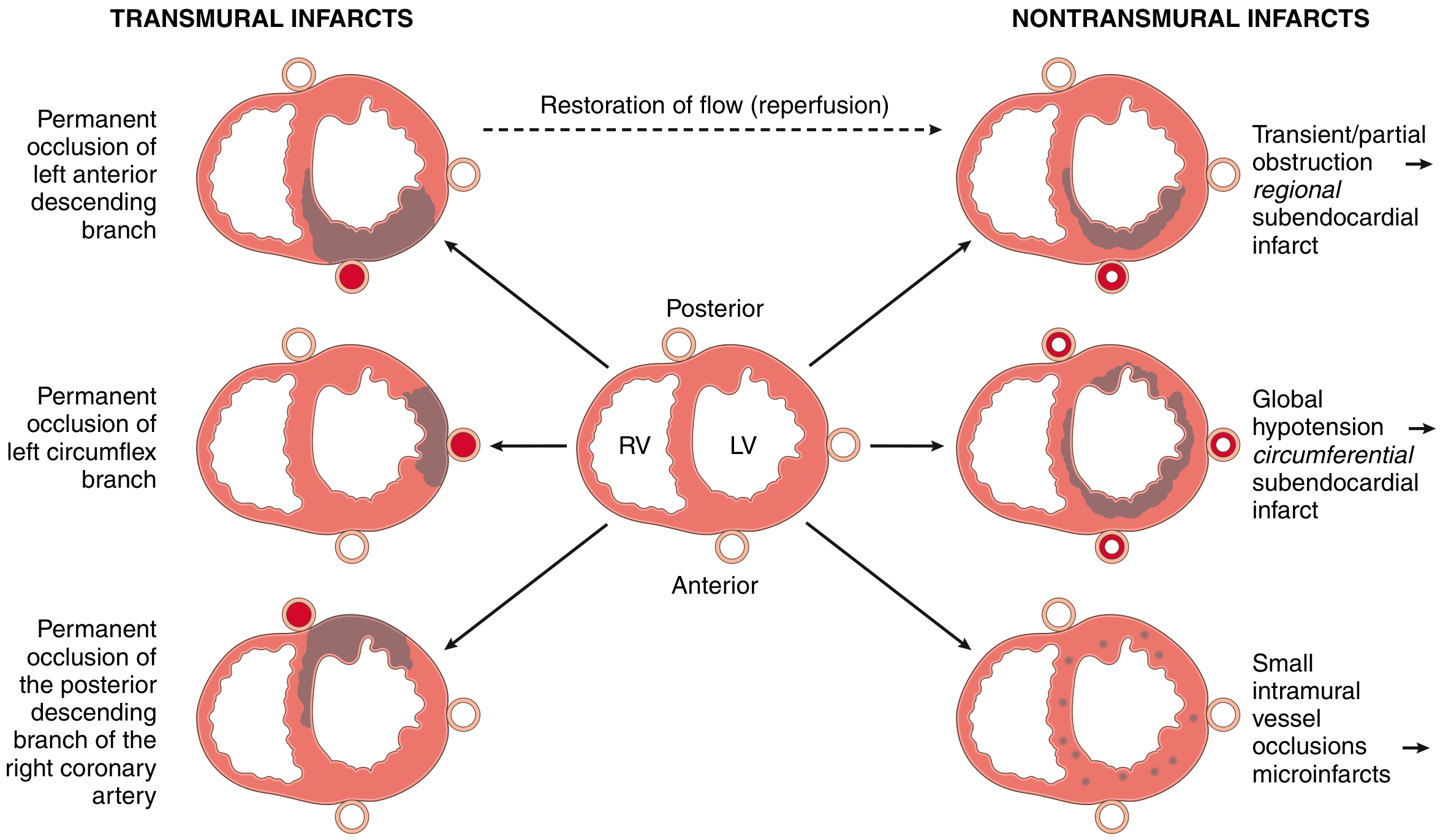
FIG. 9.9 - Left: transmural infarcts from permanent occlusion of LAD, LCX, or RCA. Right: nontransmural patterns from partial obstruction, global hypotension, or small vessel disease. (Robbins & Kumar Basic Pathology)
Key vessel territories:
- LAD occlusion (40-50% of MIs): anterior LV wall, anterior 2/3 of septum, apex
- RCA occlusion (30-40%): right ventricle, posterior/inferior LV
- LCX occlusion (15-20%): lateral LV wall
Step 6 - Morphological Evolution Over Time
Gross specimen (TTC stain):
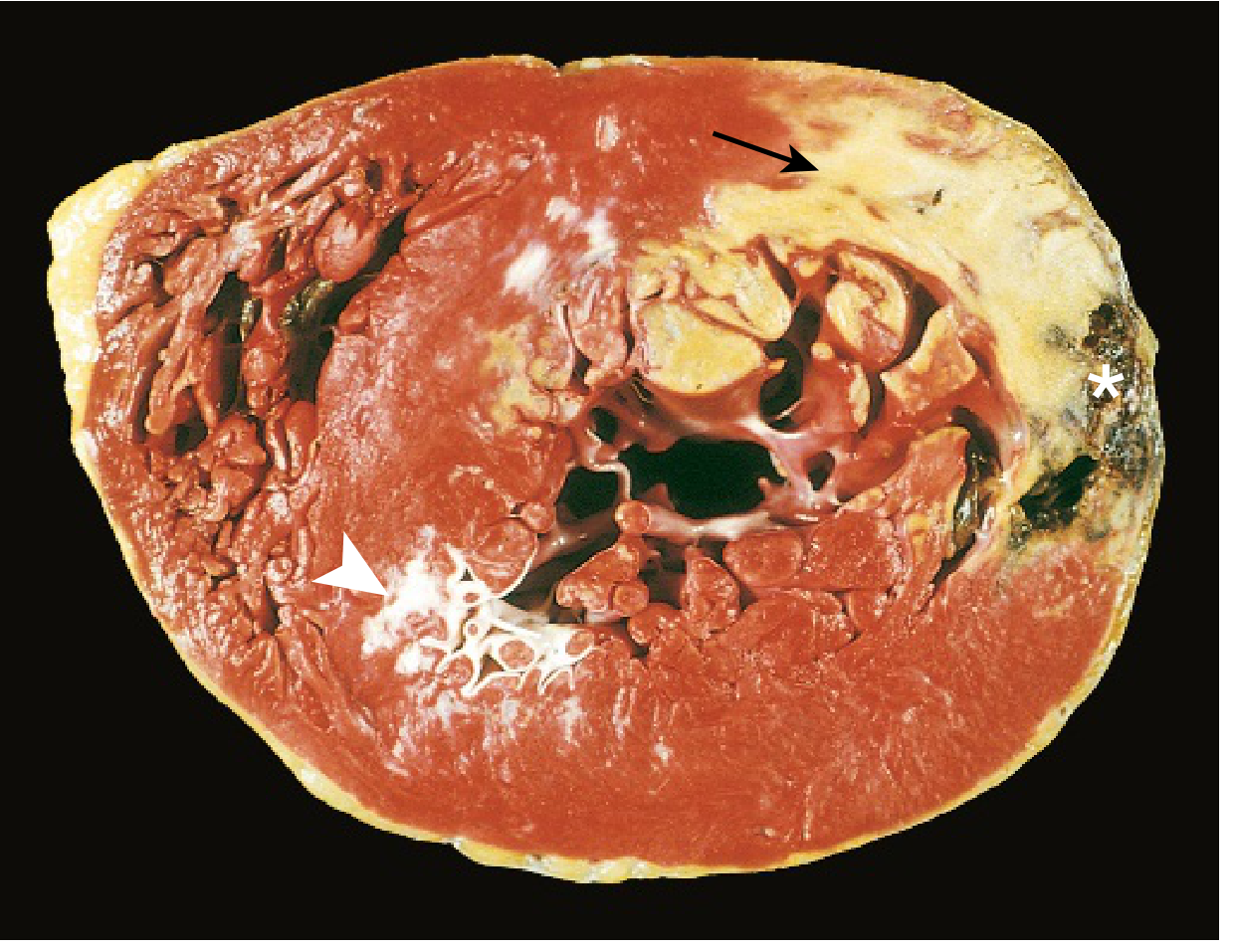
FIG. 9.10 - Acute MI of posterolateral LV: pale unstained area = necrosis (arrow); white glistening area = remote scar (arrowhead); hemorrhage at edge = ventricular rupture (). (Robbins & Kumar Basic Pathology)*
Microscopic evolution (H&E and Masson trichrome):
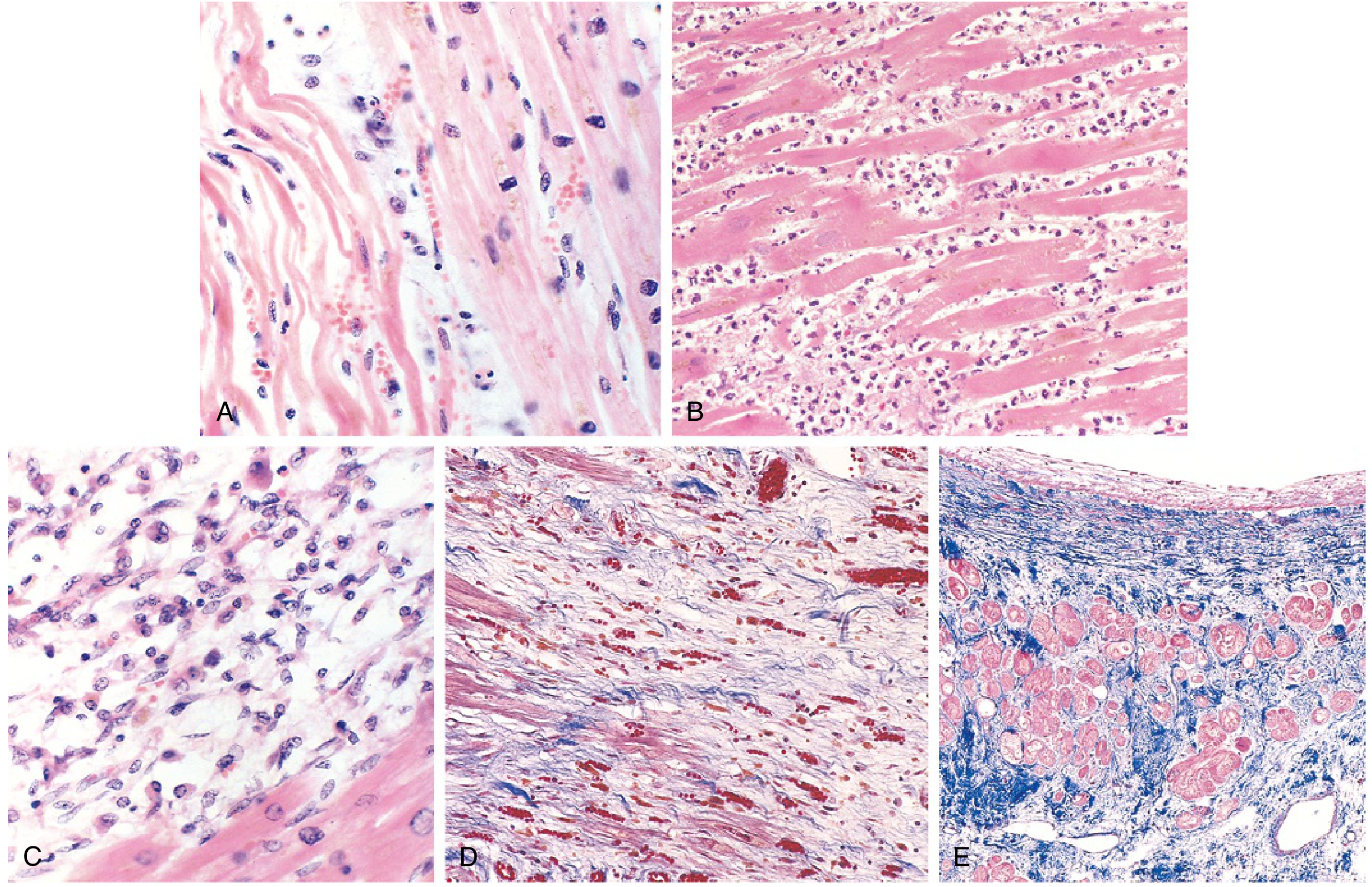
FIG. 9.11 - (A) 1-day infarct: coagulative necrosis with wavy fibers. (B) 2-3 days: dense neutrophil infiltrate. (C) 7-10 days: macrophage phagocytosis of necrotic debris. (D) Granulation tissue with new capillaries. (E) Healed scar - dense blue collagen (Masson trichrome). (Robbins & Kumar Basic Pathology)
Complete Timeline Summary
| Time Frame | Gross | Microscopy |
|---|---|---|
| 0 - 30 min | None | None (reversible: mitochondrial swelling, glycogen loss) |
| 30 min - 4 hr | None | Sarcolemmal disruption; wavy fiber change at border |
| 4-12 hr | Occasional dark mottling | Onset coagulative necrosis; edema; hemorrhage |
| 12-24 hr | Dark mottling | Coagulative necrosis; pyknotic nuclei; hypereosinophilic myocytes; early neutrophils |
| 1-3 days | Yellow-tan infarct center | Loss of nuclei/striations; dense neutrophils |
| 3-7 days | Hyperemic border; central softening | Dying neutrophils; early macrophage phagocytosis |
| 7-10 days | Maximally yellow-tan, soft | Well-developed macrophage phagocytosis; early granulation tissue |
| 2-8 weeks | Gray-white scar forming | Granulation tissue → fibrosis |
| >2 months | Dense white scar | Dense collagen scar; no regeneration |
- Robbins & Kumar Basic Pathology, Table 9.2
Reperfusion Injury
If blood flow is restored after irreversible injury has begun, reperfusion injury adds to myocyte death via:
- Massive intracellular calcium influx → uncontrolled myofibril contraction → contraction band necrosis
- Reactive oxygen species (ROS) from reperfused leukocytes damage membranes
- "No-reflow" phenomenon - leukocyte aggregation occludes the microvasculature despite opening the epicardial vessel
- Platelet and complement activation contributing to microvascular injury
Despite reperfusion injury, early reperfusion (within the 3-6 hour window) is the single most effective intervention to limit infarct size.
MI Without Atherothrombosis (~10% of cases)
-
Vasospasm (cocaine, ephedrine use; Prinzmetal angina)
-
Embolism from atrial fibrillation mural thrombus, valve vegetations, or intracardiac prostheses
-
Vasculitis, amyloid deposition, or sickle cell vaso-occlusion of small intramyocardial arterioles
-
Demand ischemia: severe fixed coronary disease + tachycardia, hypertension, or anemia
-
Robbins & Kumar Basic Pathology; Robbins Cotran & Kumar Pathologic Basis of Disease
This is a shared conversation. Sign in to Orris to start your own chat.