For the topic of pathology cardiomyopathy
Asking for Preferences
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
Loading Image
I have comprehensive content and all three pathology images. Now I'll compile the complete study notes.
Pathology of Cardiomyopathy — Study Notes
Sources: Robbins & Kumar Basic Pathology (Robbins Pathology), Robbins Cotran & Kumar Pathologic Basis of Disease, Goldman-Cecil Medicine, Braunwald's Heart Disease
Overview
Cardiomyopathies are intrinsic cardiac muscle diseases that may be genetic or due to other well-defined causes. They are distinct from myocardial dysfunction secondary to ischaemic heart disease, valvular disease, hypertension, or congenital defects.
Three pathophysiologic categories:
| Type | Frequency | Core Dysfunction | LVEF |
|---|---|---|---|
| Dilated (DCM) | ~90% | Systolic (contractile) dysfunction | < 40% |
| Hypertrophic (HCM) | Less common | Diastolic (relaxation) dysfunction | 50–80% |
| Restrictive (RCM) | Least common | Diastolic (compliance) dysfunction | 25–50% |
(ARVC and Takotsubo/Stress are additional recognised forms)
— Robbins & Kumar Basic Pathology, p. 371 (Table 9.5)
1. Dilated Cardiomyopathy (DCM)
Definition & Epidemiology
Progressive cardiac dilation + contractile (systolic) dysfunction, usually with concurrent hypertrophy. The most common cardiomyopathy (~90% of cases).
Pathogenesis
Genetic causes (20–50% of cases)
-
50 causative genes identified; autosomal dominant predominant
- Loss-of-function mutations in cytoskeletal/sarcomeric proteins
- Most common gene: TTN (titin) — truncating mutations in ~20–25% of familial DCM and 18% of sporadic cases
- Other genes: β-myosin heavy chain, α-myosin heavy chain, cardiac troponin T, desmin (intermediate filament), nuclear lamins A/C
- Lamin A/C mutations → arrhythmia + progressive AV conduction disease preceding DCM
- X-linked DCM: dystrophin gene mutations (also causes Duchenne/Becker MD); accounts for 2–5% of familial cases
⚡ Key contrast: Gain-of-function mutations in sarcomere genes (β-MHC) → HCM; Loss-of-function mutations in same genes → DCM
Acquired causes
- Viral myocarditis → coxsackievirus B, parvovirus B19, HHV-6, adenovirus; often leaves viral "footprints" in end-stage DCM
- Alcohol/toxins: Alcohol + acetaldehyde direct cardiotoxicity → myocyte vacuolisation, mitochondrial abnormalities, fibrosis. Estimated threshold: ~6 drinks/day for 5–10 years. Abstinence improves >50%
- Chemotherapy: Anthracyclines (doxorubicin, daunorubicin) — dose-dependent; risk at >450 mg/m²
- Pregnancy (peripartum cardiomyopathy)
- Iron overload, radiation, nutritional deficiency, cocaine, sarcoidosis, tachycardia-mediated
Morphology (Gross & Microscopic)
Gross: Four-chamber dilation and hypertrophy. Flabby, poorly contractile walls. Mural thrombus commonly at the LV apex (risk of embolism).
Microscopic (Masson trichrome): Myocyte hypertrophy + interstitial fibrosis (blue collagen on trichrome). Non-specific; no pathognomonic features at end stage.
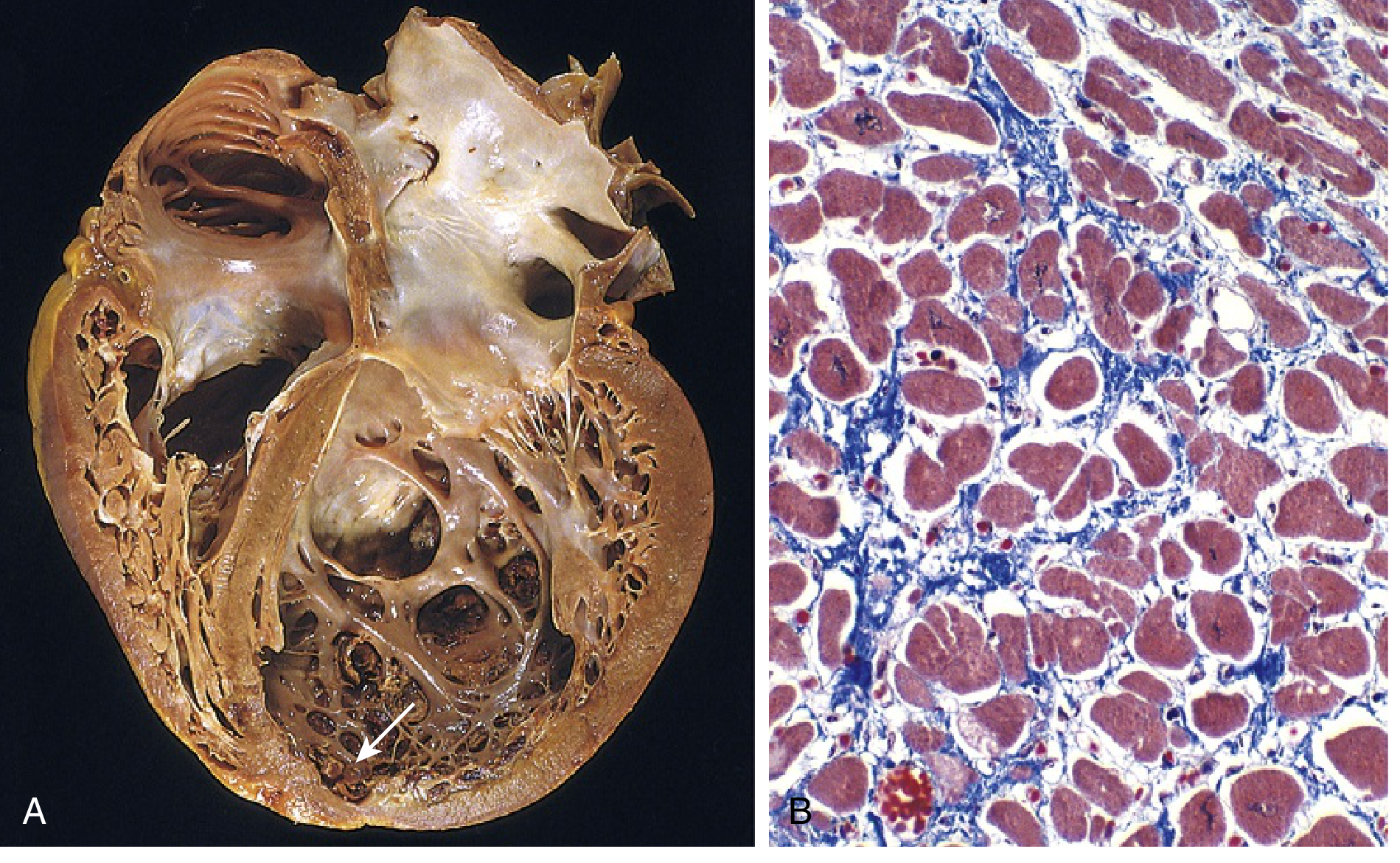
Clinical Features
- Symptoms of high pulmonary venous pressure (dyspnoea, orthopnoea, PND) or low cardiac output
- Palpitations, presyncope, syncope (arrhythmias)
- Advanced: sinus tachycardia, weak pulses, hypotension, elevated JVP, displaced apex
- Complications: CHF, systemic/pulmonary emboli from mural thrombi, sudden cardiac death, AF
Management Principles
Heart failure therapy (ACE-I/ARB, beta-blockers, diuretics, aldosterone antagonists); ICD for arrhythmia; cardiac resynchronisation therapy; transplant in refractory cases.
2. Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)
Definition
Autosomal dominant disorder characterised by right-sided heart failure and life-threatening ventricular arrhythmias, with a strong predilection for sudden cardiac death (SCD) in young athletes.
Epidemiology
- Prevalence: 1 in 2000–5000 adults
- Accounts for ~10% of sudden cardiac deaths in athletes
Pathogenesis
- Mutations in desmosomal proteins at the intercalated disk — most commonly plakoglobin, plakophilin-2, desmoplakin, desmoglein-2, desmocollin-2
- Also mutations in desmin (intermediate filament interacting with desmosome)
- Mechanism: Desmosomal detachment → myocyte death (especially during strenuous exercise) → progressive replacement by fat and fibrosis
Morphology
Gross: Severely thinned RV free wall, markedly dilated RV. LV may be involved to a lesser extent in some cases.
Microscopic (Masson trichrome): RV myocardium focally replaced by fibro-fatty tissue — adipose tissue (white) and fibrous connective tissue (blue on trichrome).
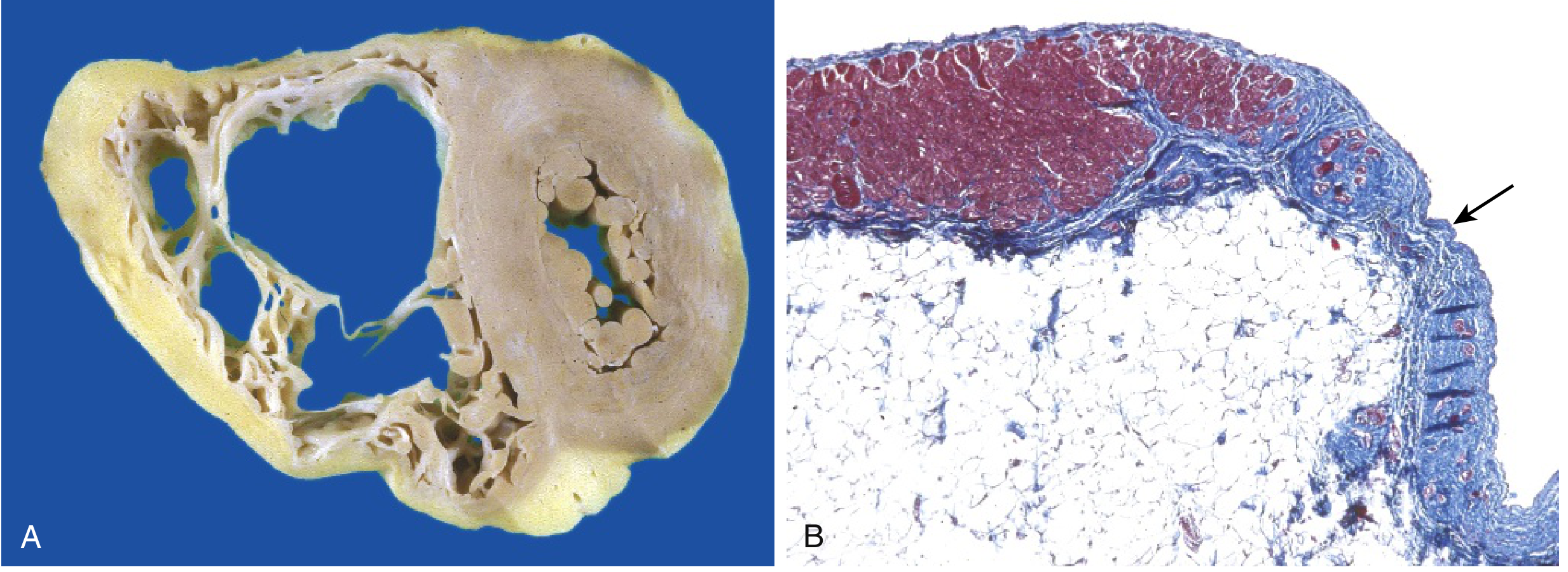
Clinical Features
- Palpitations, syncope, sudden cardiac death (often first presentation)
- Ventricular tachycardia with LBBB morphology (origin in RV)
- ECG: epsilon waves, T-wave inversions in V1–V3
- Diagnosis: Task Force Criteria (imaging, ECG, histology, genetics, family history)
3. Hypertrophic Cardiomyopathy (HCM)
Definition
Massive myocardial hypertrophy without ventricular dilation, with preserved or hyperdynamic systolic function, but impaired diastolic filling. One-third have outflow tract obstruction (HOCM).
Pathogenesis
- Gain-of-function mutations in sarcomeric proteins → hypercontractility → increased energy consumption → net negative energy balance
- Autosomal dominant; >400 causative mutations in 9 genes
- Top 3 genes (account for 70–80% of HCM):
- β-myosin heavy chain (MYH7) — most frequently involved
- Myosin-binding protein C (MYBPC3)
- Cardiac troponin T (TNNT2)
- Same genes as DCM but mutations cause gain vs loss of function
Morphology
Gross:
- Massive myocardial hypertrophy without ventricular dilation
- 90%: Asymmetric septal hypertrophy (ASH) — septum thicker than LV free wall
- Ventricular cavity → "banana-shaped" configuration on longitudinal section
- Anterior mitral leaflet contacts the hypertrophied septum during systole → fibrous endocardial plaque in LVOT + SAM (systolic anterior motion)
- Both atria dilated
Microscopic (H&E):
- Marked myocyte hypertrophy
- Myocyte/myofiber disarray — haphazard arrangement (pathognomonic)
- Interstitial fibrosis
- Characteristic branching of myocytes
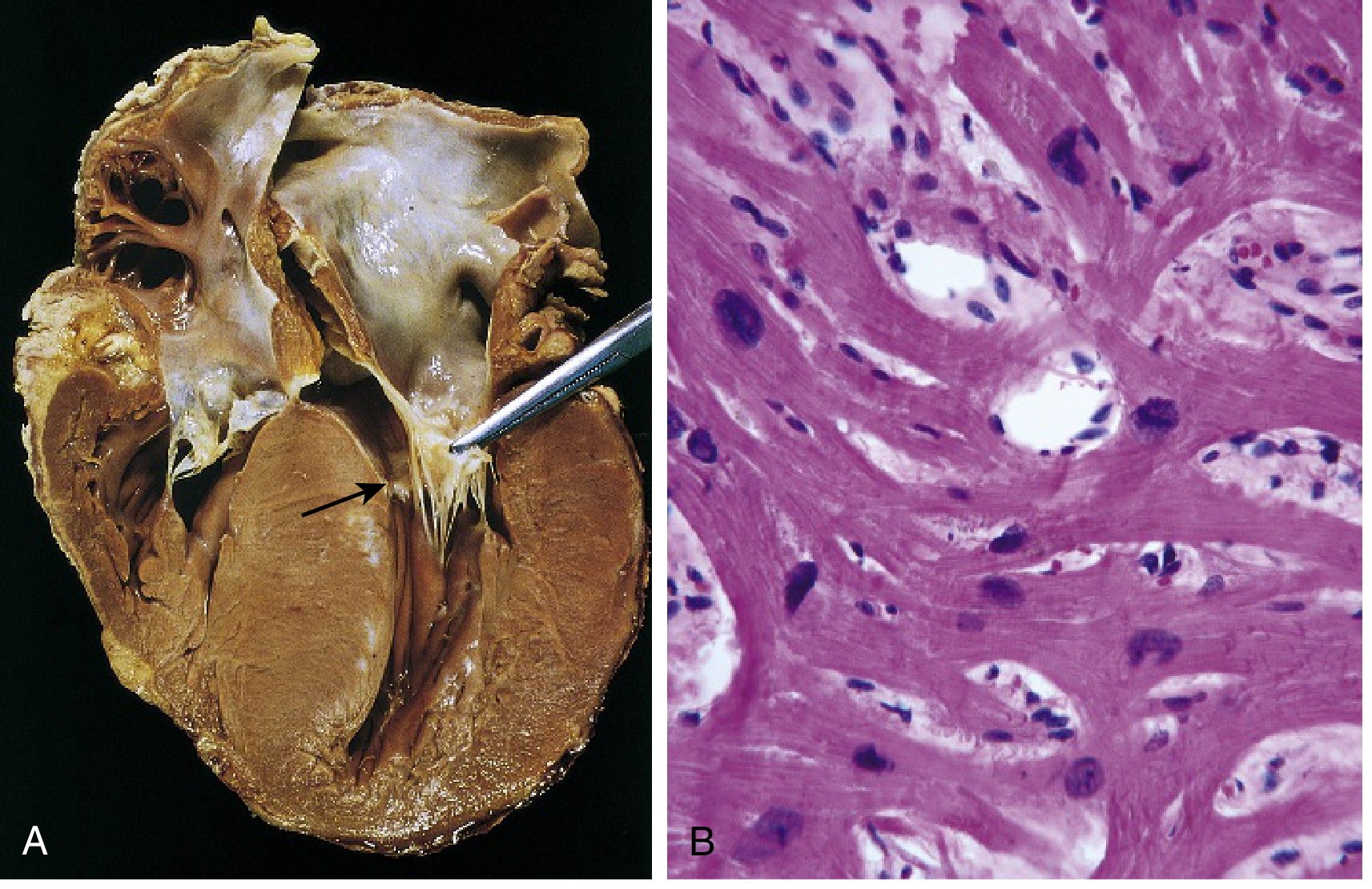
Clinical Features
- Typically manifests at postpubertal growth spurt
- Exertional dyspnoea + harsh systolic ejection murmur (outflow obstruction)
- Angina — due to massive hypertrophy + increased O₂ demand + compromised intramural coronary flow (even without CAD)
- Murmur increases with Valsalva/standing (↓ preload → more obstruction)
- Murmur decreases with squatting/leg raise (↑ preload)
Major Complications
| Complication | Mechanism |
|---|---|
| Sudden cardiac death | Ventricular fibrillation — #1 cause of SCD in athletes <35 years (1/3 of cases) |
| Atrial fibrillation | Left atrial dilation; risk of mural thrombus & stroke |
| Infective endocarditis | Mitral valve (turbulent flow) |
| Congestive heart failure | Chronic diastolic dysfunction |
Management
- Beta-blockers or calcium channel blockers (promote ventricular relaxation)
- Avoid vasodilators, dehydration, intense exercise
- ICD for high-risk SCD
- Septal reduction: surgical myectomy or alcohol septal ablation
- Mavacamten (myosin inhibitor) — reduces hypercontractility
4. Restrictive Cardiomyopathy (RCM)
Definition
Decreased ventricular compliance → impaired ventricular filling during diastole ("stiff ventricle"). The least common primary cardiomyopathy.
Pathogenesis & Causes
Primary:
- Idiopathic (interstitial fibrosis)
Secondary (infiltrative/storage):
| Cause | Key Feature |
|---|---|
| Amyloidosis (most common systemic cause) | AL type (light chains) or ATTR (transthyretin); ATTR common in elderly Black men (Val122Ile mutation) |
| Sarcoidosis | Non-caseating granulomas; also causes conduction defects and VT |
| Haemochromatosis | Iron deposition; responds to phlebotomy/chelation |
| Glycogen storage diseases | Pompe disease, Fabry disease |
| Radiation fibrosis | Post-radiotherapy interstitial fibrosis |
| Mucopolysaccharidoses | Hurler, Hunter syndromes |
Endomyocardial forms:
- Endomyocardial fibrosis: Most common worldwide (tropical Africa); diffuse fibrosis of ventricular endocardium + sub-endocardium; affects tricuspid and mitral valves; linked to helminthic infections and nutritional deficiency
- Loeffler endomyocarditis: Peripheral hypereosinophilia + eosinophilic tissue infiltrates; eosinophil granule products (major basic protein) → endocardial/myocardial necrosis → scarring → mural thrombus
Morphology
- Gross: Ventricles of approximately normal size, cavities not dilated, myocardium firm; both atria typically dilated (due to restricted ventricular filling and pressure overload)
- Microscopic: Variable interstitial fibrosis; endomyocardial biopsy reveals specific cause (amyloid deposits, granulomas, iron, eosinophilic infiltrates)
Clinical Features
- Dyspnoea, fatigue, signs of right-sided failure (elevated JVP, peripheral oedema, ascites)
- Heart sounds may be normal; S3/S4 gallop
- ECG: Low voltage (especially in amyloid), conduction abnormalities
- Echo: Normal or mildly reduced systolic function, biatrial enlargement, diastolic dysfunction
- Key distinction from constrictive pericarditis (can be clinically identical — requires cardiac catheterisation or CMR)
Congo Red Staining in Amyloidosis
- Amyloid deposits stain orange-red with Congo red; show apple-green birefringence under polarised light
5. Stress (Takotsubo) Cardiomyopathy
Definition
Transient LV dysfunction, characterised by apical ballooning and basal hypercontractility, typically triggered by emotional or physical stress. Also known as "broken heart syndrome," "apical ballooning syndrome," or "ampulla cardiomyopathy."
Epidemiology
- Accounts for ~4% of patients presenting with presumed ACS
- ~90% female, predominantly postmenopausal (prevalence 5.9–7.5% in women presenting with ACS)
Pathogenesis
- Trigger: Acute emotional/physical stress → massive catecholamine surge
- Catecholamines cause direct myocardial toxicity (catecholamine-mediated microvascular spasm) or bind β-receptors in the apex (which has higher β-receptor density) → apical stunning
- The 4th Universal Definition of MI does not classify Takotsubo as AMI — it is recognised as a separate syndrome, not MINOCA
Morphology / Findings
- LV apical ballooning (akinesis/dyskinesis of apex + mid-ventricle)
- Hypercontractile LV base
- On CMR: transient wall motion abnormality; can show mid-wall fibrosis in some cases
- Gross pathology: Non-specific; no permanent structural damage in typical cases
Clinical Features
- Presents identically to STEMI: chest pain, ST elevation/T-wave changes, mild troponin rise
- Precipitated by sudden emotional shock (grief, fear, anger) or physical stressor (surgery, stroke, sepsis)
- Coronary angiography: No obstructive CAD (key distinguishing feature)
- Usually reversible — LV function recovers within weeks
Prognosis & Complications
- Not purely benign: in-hospital mortality 4.1%, long-term (1-year) mortality 5.6% (InterTAK Registry)
- Recurrence in 5–10%
- Complications: Acute heart failure, LV outflow obstruction (due to basal hypercontractility), cardiogenic shock, ventricular arrhythmias, LV thrombus, mitral regurgitation
Management
- Supportive care; treat precipitant
- ACE-I — observational benefit; improved 1-year survival
- Beta-blockers — no proven benefit for index event or prevention of recurrence
- No randomised controlled trials guide therapy
Quick Comparison Table
| Feature | DCM | ARVC | HCM | RCM | Takotsubo |
|---|---|---|---|---|---|
| LV wall | Thinned, dilated | Normal LV (RV affected) | Thickened, non-dilated | Normal/slightly enlarged | Apical akinesis |
| Systolic fn | ↓↓ (EF <40%) | RV failure | Preserved/hyperdynamic | Mildly ↓ | Transient ↓ (apex) |
| Diastolic fn | Normal/mildly ↓ | Normal | ↓↓ | ↓↓ | Transient ↓ |
| Genetics | Loss-of-function (TTN, MYH7, LMNA) | Desmosomal proteins (plakoglobin) | Gain-of-function (MYH7, MYBPC3, TNNT2) | Variable (ATTR amyloid) | None |
| Key micro | Myocyte hypertrophy + fibrosis | Fibrofatty RV replacement | Myocyte disarray + fibrosis | Interstitial fibrosis + specific deposits | Non-specific (transient) |
| SCD risk | Yes (VF) | High (athletes) | High — #1 in athletes <35 yr | Low | Low |
| Key complication | CHF, embolism | SCD, RV failure | SCD, AF, endocarditis | CHF, AF | Reversible; ACS mimic |
| Murmur | MR (functional) | None | Harsh systolic ejection | None | Dynamic LVOTO possible |
Key Pathology Pearls for Exams
- DCM vs HCM genetics: Both can involve β-MHC and troponin T mutations — but DCM = loss of function; HCM = gain of function
- HCM hallmark histo: Myocyte disarray — random, haphazard orientation of myofibers with bizarre branching
- ARVC hallmark histo: Fibrofatty replacement of RV wall (Masson trichrome: blue fibrosis + white fat replacing red myocardium)
- RCM with Congo red stain → apple-green birefringence = amyloid
- Endomyocardial fibrosis = most common RCM worldwide (tropical areas); Loeffler = eosinophilia-associated
- Takotsubo = no obstructive CAD on angiography + stress trigger + women 90% + reversible
- Titin (TTN) = most common single gene mutated in DCM
- MYH7 (β-myosin heavy chain) = most common gene mutated in HCM
- ARVC = desmosomal protein mutations — avoid strenuous exercise; SCD risk in athletes
- "Banana-shaped" LV lumen = HCM with ASH
- Atrial dilation without ventricular dilation = hallmark gross finding in RCM
- Mural thrombus at LV apex = DCM (due to stasis in dilated poorly contractile ventricle)
Sources: Robbins & Kumar Basic Pathology — pp. 371–375 (Table 9.5, Figs 9.25–9.27) | Goldman-Cecil Medicine — Chapter 47 | Braunwald's Heart Disease — Chapter on Sex Differences in CVD | Robbins Cotran & Kumar Pathologic Basis of Disease — p. 533
This is a shared conversation. Sign in to Orris to start your own chat.