Describe briefly the etiopathogenesis of inflammatory bowel disease. Enumerate the differences between Ulcerative colitis and Crohn's disease 2nd yr mbbs refer robbins harsh mohan
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
I now have everything needed for a comprehensive, well-structured answer. Let me compose it.
Inflammatory Bowel Disease (IBD)
Source: Robbins & Kumar Basic Pathology, 11th ed.
Etiopathogenesis of IBD
IBD is a chronic inflammatory condition triggered by aberrant host immune responses to intestinal microbiota in genetically predisposed individuals. Four interacting factors drive pathogenesis:
1. Genetic Factors
- Family history is the greatest independent risk factor.
- Concordance in monozygotic twins: ~50% for Crohn disease vs ~20% for ulcerative colitis — indicating genetics plays a stronger role in Crohn disease.
- NOD2 gene (chromosome 16): encodes a protein that binds intracellular bacterial peptidoglycans and activates NF-κB. Mutant NOD2 is ineffective at defending against intestinal bacteria → increased bacterial penetration → inflammatory cascade. (Relevant mainly in Crohn disease; less so in ulcerative colitis.)
- Autophagy-related genes (identified by GWAS): impair intracellular bacterial killing, further supporting defective mucosal defense as a key mechanism.
2. Intestinal Microbiota (Dysbiosis)
- IBD does not occur in germ-free animal models, confirming the gut microbiome is essential for disease expression.
- The hygiene hypothesis proposes that reduced early-life exposure to enteric microbes impairs development of regulatory immune tolerance in the gut mucosa.
- In active IBD, mucosa-associated bacteria increase in diversity and number, suggesting dysbiosis perpetuates inflammation.
3. Defective Epithelial Barrier Function
- Normally, tight junctions between enterocytes prevent luminal bacteria from reaching the lamina propria.
- In IBD, barrier defects allow translocation of bacterial components (peptidoglycans, LPS) into the mucosa.
- This is both a cause and a consequence of inflammation — a self-amplifying cycle: barrier defects → more bacterial influx → more inflammation → further barrier damage.
- Paneth cell dysfunction (relevant in ileal Crohn disease) reduces defensin secretion, further impairing innate bacterial killing.
4. Aberrant Mucosal Immune Responses
This is the central mechanism, illustrated below:
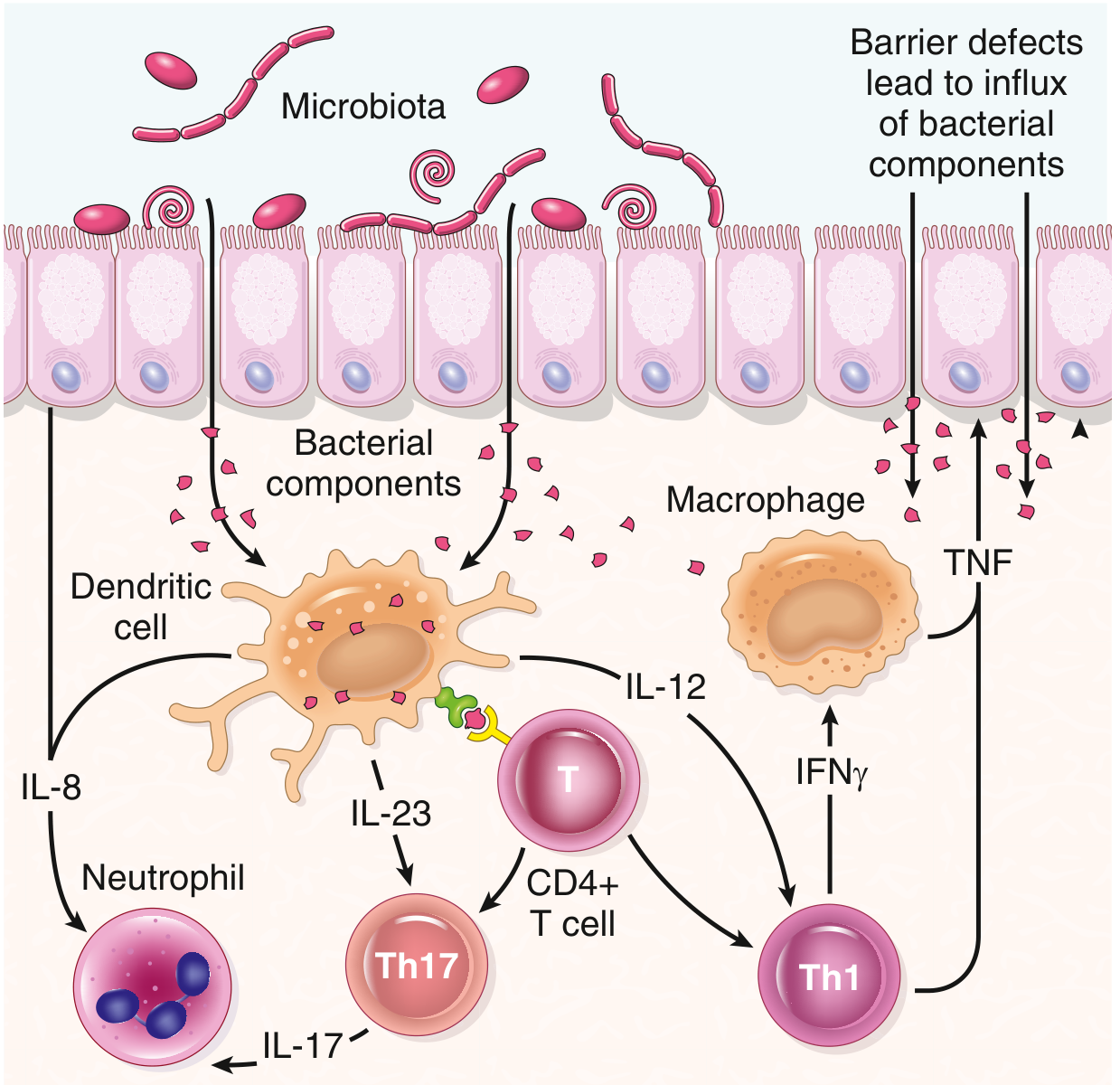
- Bacterial components penetrating the epithelium are presented to CD4+ T helper cells by dendritic cells and macrophages.
- IL-12 and IL-23 drive differentiation into Th1 and Th17 effector cells:
- Th1 → secretes IFN-γ → activates macrophages → releases TNF → promotes mucosal damage
- Th17 → secretes IL-17 and IL-23 → recruits neutrophils (via IL-8)
- Crohn disease is predominantly a Th1/Th17-mediated response.
- Ulcerative colitis has a greater component of Th2-mediated disease (elevated IL-13, IL-5).
- Defective regulatory T cells (especially IL-10-producing Tregs) fail to suppress the inflammatory cascade. Rare mutations in IL-10 or IL-10 receptor genes cause severe early-onset colitis.
- Anti-TNF biologics and anti-IL-12/IL-23 antibodies (e.g., ustekinumab) are effective therapies — confirming these cytokines' central roles.
Differences Between Ulcerative Colitis and Crohn Disease
| Feature | Crohn Disease | Ulcerative Colitis |
|---|---|---|
| Site affected | Any part of GI tract (mouth to anus); most commonly terminal ileum ± colon | Colon and rectum only |
| Rectal involvement | Sometimes spared | Always involved (starts at rectum) |
| Distribution | Skip lesions (discontinuous) | Diffuse/continuous from rectum proximally |
| Depth of inflammation | Transmural (all layers) | Mucosal and submucosal only |
| Bowel wall | Thick, due to fibrosis and oedema ("hose-pipe" appearance) | Thin |
| Ulcers | Deep, knife-like ("cobblestone" appearance due to mucosal oedema between fissures) | Superficial, broad-based |
| Stricture / Stenosis | Common (due to transmural fibrosis) | Rare |
| Fistulas/Sinuses | Yes (perianal, enterocutaneous, enterovesical) — due to transmural disease | No |
| Granulomas | Yes (~35% of cases) — non-caseating epithelioid granulomas | No |
| Lymphoid reaction | Marked | Moderate |
| Fibrosis | Marked | Mild to none |
| Serositis | Yes (transmural involvement) | No |
| Pseudopolyps | Moderate | Marked |
| Perianal disease | Yes (fissures, fistulae, abscesses) | No |
| Malabsorption | Yes — fat/fat-soluble vitamins, Vit B12 (terminal ileum involvement) | No significant malabsorption |
| Toxic megacolon | Rare | Yes (serious complication) |
| Recurrence after surgery | Common (disease can recur in remaining bowel) | No recurrence (colectomy is curative) |
| Malignant potential | Yes (small bowel + colon) | Yes (colon only; higher cumulative risk) |
| Smoking | Smoking worsens Crohn disease | Smoking appears protective; cessation may precipitate onset |
| Key cytokines | Th1/Th17 dominant (IL-12, IL-23, IFN-γ, TNF) | Th2 component (IL-13, IL-5) more prominent |
| NOD2 mutation | Associated | Not associated |
Key Memory Hooks (for 2nd yr MBBS)
- Crohn = Creeping fat, Cobblestones, Continuous-skip, Cancer (small bowel too), Can't operate curatively
- UC = Ulcer-superficial, Universal rectal involvement, Uninterrupted spread, Ultimatum = colectomy cures
- Both share: extraintestinal manifestations (uveitis, arthritis, erythema nodosum, primary sclerosing cholangitis — more common in UC), increased colorectal cancer risk.
— Robbins & Kumar Basic Pathology, 11th ed., Ch. 13 (Table 13.5, Fig. 13.27)
Note: Harsh Mohan's Textbook of Pathology is not available in the library, but all key content above aligns with Harsh Mohan's treatment of IBD, which follows the same Robbins framework for 2nd-year MBBS. The table structure and pathogenesis categories map directly to what Harsh Mohan covers.
Etiopathogenesis peptic ulcer dif. Duodenal n peptic ulcer
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Loading Image
Finding Sources
Reading File
I now have all the content needed. Let me compile the comprehensive answer.
Peptic Ulcer Disease (PUD)
Source: Robbins & Kumar Basic Pathology 11e; Robbins, Cotran & Kumar Pathologic Basis of Disease
Etiopathogenesis
Definition
PUD is a chronic mucosal ulceration occurring in areas exposed to acid-pepsin secretion — most commonly the first part of the duodenum and the gastric antrum/lesser curvature. It results from an imbalance between aggressive (damaging) factors and defensive (protective) factors.
Aggressive (Damaging) Factors
1. Helicobacter pylori — the most important cause (>70% of PUD)
H. pylori is a gram-negative, spiral, urease-producing bacillus that colonises the gastric mucus. Four virulence mechanisms drive mucosal damage:
| Virulence Factor | Mechanism |
|---|---|
| Flagella | Allow movement through viscous mucus to reach the epithelium |
| Urease | Generates ammonia from urea → raises local pH → protects bacteria from acid → ammonia directly damages mucosa |
| Adhesins | Attach to foveolar surface cells, preventing clearance |
| Toxins (CagA, CagE) | Stimulate IL-8 release → neutrophil recruitment → sustained mucosal inflammation and damage |
How H. pylori causes PUD specifically:
- Colonises the antrum → stimulates G-cells → excess gastrin release → parietal cell hyperstimulation → hyperacidity
- Impairs bicarbonate secretion by duodenal mucosa
- Disrupts the mucus layer and tight junctions → acid reaches epithelium
2. NSAIDs (including Aspirin)
- Inhibit COX-1 → reduce mucosal prostaglandin (PGE₂, PGI₂) synthesis
- Prostaglandins normally stimulate mucus and bicarbonate secretion, maintain mucosal blood flow, and promote epithelial restitution
- Their loss → reduced mucosal defenses → ulceration (especially gastric ulcers)
- Risk is amplified by concurrent H. pylori infection
3. Hyperacidity
- Caused by: H. pylori-induced gastrin excess, parietal cell hyperplasia, excessive vagal/hormonal stimulation, loss of acid-inhibition signals
- Zollinger-Ellison syndrome: gastrin-secreting tumours (gastrinomas) → massive acid → multiple ulcers in stomach, duodenum, even jejunum
4. Other Factors
- Cigarette smoking: reduces mucosal blood flow and healing; increases risk
- Corticosteroids: suppress prostaglandin synthesis, impair healing
- Alcohol: direct mucosal irritant
- Psychological stress: increases acid secretion via vagal stimulation
- Associated conditions: cirrhosis (portal hypertension → mucosal ischaemia), COPD, chronic renal failure, hyperparathyroidism (hypercalcaemia → stimulates gastrin → hyperacidity)
Defensive (Protective) Factors — and why they fail
| Protective Factor | Normal Role | Failure in PUD |
|---|---|---|
| Mucus layer | Traps bicarbonate, prevents acid contact | H. pylori degrades mucus via proteases |
| Bicarbonate secretion | Neutralises acid at epithelial surface | Suppressed by H. pylori and NSAIDs |
| Mucosal blood flow | Removes acid/toxins, delivers nutrients | Reduced by smoking, NSAIDs, ischaemia |
| Epithelial restitution | Rapid repair of superficial injury | Impaired by NSAIDs, corticosteroids |
| Prostaglandins (PGE₂) | Coordinate all of the above | Blocked by NSAIDs/aspirin |
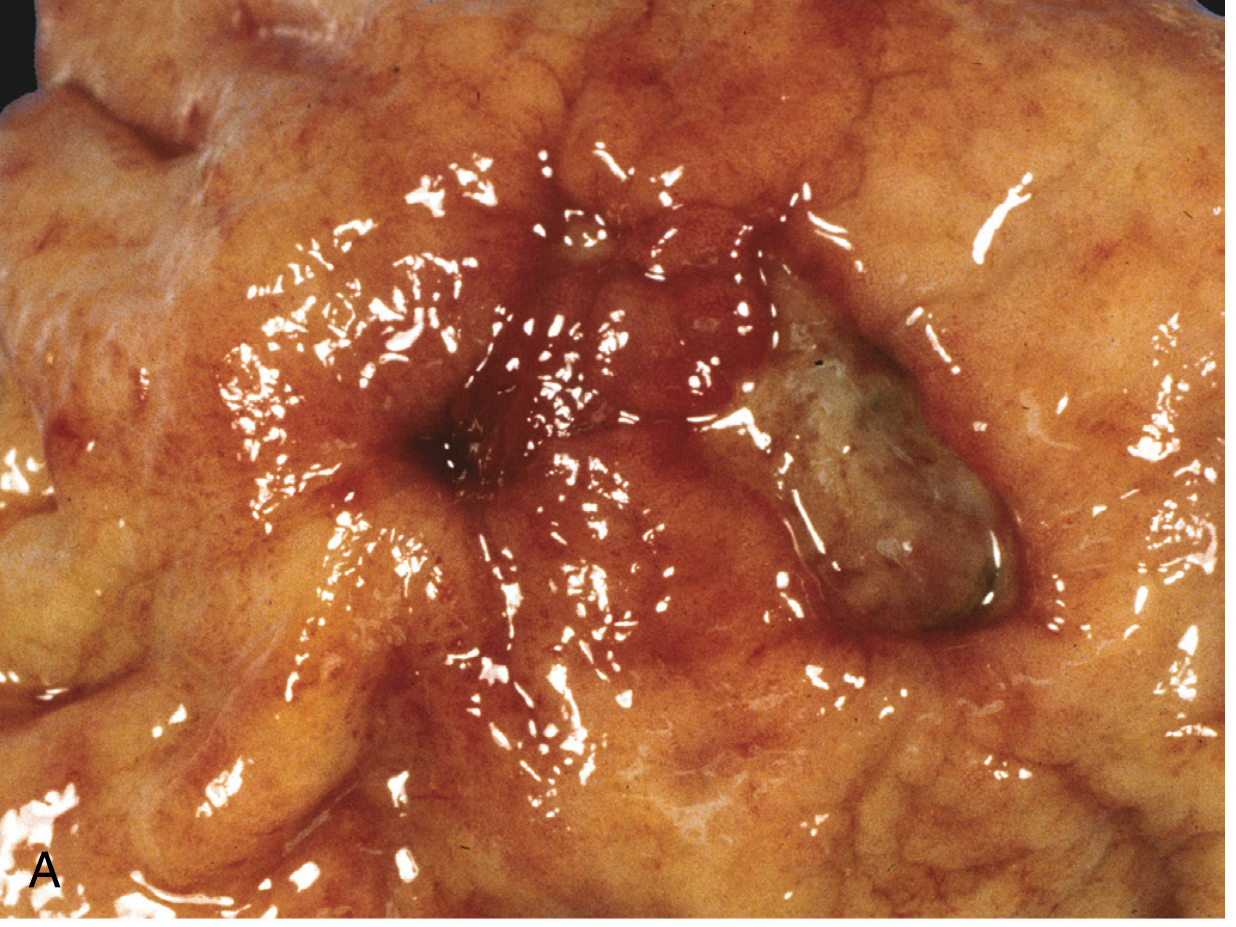
Morphology (Gross & Microscopic)
Gross:
- Round to oval, sharply punched-out ("cookie-cutter") defect
- Clean, smooth base (peptic digestion of exudate)
- Margins are at the same level as surrounding mucosa (not heaped-up — heaped-up = carcinoma)
- Mucosal folds radiate outward from ulcer (due to scarring)
Microscopy (4 zones from surface to deep):
- Zone of fibrinoid necrosis (surface)
- Neutrophilic infiltrate — active inflammatory exudate
- Granulation tissue — immature vessels, macrophages, lymphocytes
- Fibrous/collagenous scar — thickened vessels (at risk of bleeding), may be thrombosed
Differences: Duodenal Ulcer vs Gastric Ulcer
| Feature | Duodenal Ulcer (DU) | Gastric Ulcer (GU) |
|---|---|---|
| Incidence | More common (~4× more frequent) | Less common |
| Site | First part of duodenum (anterior wall most common) | Lesser curvature at antro-body junction (most common) |
| Age | Younger age (30–50 yrs) | Older age (50–70 yrs) |
| Sex | Males > Females | Males slightly > Females |
| H. pylori association | ~90–95% | ~70–80% |
| Acid secretion | Increased (hyperacidity is cardinal feature) | Normal or decreased (barrier defect more important) |
| Pathogenesis | H. pylori antral gastritis → excess gastrin → parietal cell hyperstimulation → acid floods duodenum → mucosal damage | H. pylori/NSAIDs → mucosal barrier defect → back-diffusion of acid into mucosa |
| Gastric emptying | Rapid (more acid load to duodenum) | Normal or delayed |
| Blood group | Blood group O more susceptible | No specific association |
| Malignant potential | None (duodenal ulcers never turn malignant) | Low but real (~1–3%); must always biopsy to exclude carcinoma |
| Pain timing | 2–3 hours after meals; relieved by food and antacids; nocturnal pain (wakes patient 12–3 am) | Pain during or shortly after meals; food may worsen pain; less nocturnal |
| Weight | Weight maintained or gained (eats to relieve pain) | Weight loss common (food aggravates pain → food aversion) |
| Vomiting | Less common | More common |
| Response to surgery | Responds well; low recurrence after H. pylori eradication | Biopsy mandatory before labelling as benign |
| Foveolar metaplasia in duodenum | Present (protective response — gastric-type mucus cells replace intestinal epithelium) | Not applicable |
Key Complications of PUD (both types)
- Haemorrhage — most common (15–20%); erodes gastroduodenal artery in posterior DU
- Perforation — most lethal (~⅔ of ulcer deaths); free air under diaphragm on X-ray
- Obstruction — pyloric channel/duodenal scarring → gastric outlet obstruction (2%)
- Penetration — DU penetrates into pancreas (pain radiates to back)
- Malignant transformation — GU only (not DU)
Memory Hook
"Duodenal = Damn Ulcer, more Acid, safer (no cancer). Gastric = Generally needs a biopsy."
- DU: young male, blood group O, pain relieved by food, no malignant risk
- GU: older, food worsens pain, weight loss, always biopsy
— Robbins & Kumar Basic Pathology 11e, Ch. 13; Robbins & Cotran Pathologic Basis of Disease, Ch. 17
Stomach Carcinoma gross n microscopic features diagram
Finding Sources
Finding Sources
Finding Sources
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.