Neuromuscular junction and trnasmission along with blockers drugs toxins and diseases
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
I now have everything needed to write a comprehensive, well-illustrated answer. Let me compile it:
Neuromuscular Junction: Structure, Transmission, Blockers, Toxins & Diseases
1. Structure of the Neuromuscular Junction
The neuromuscular junction (NMJ) is the specialized synapse between a motor neuron and a skeletal muscle fiber. Key structural elements:
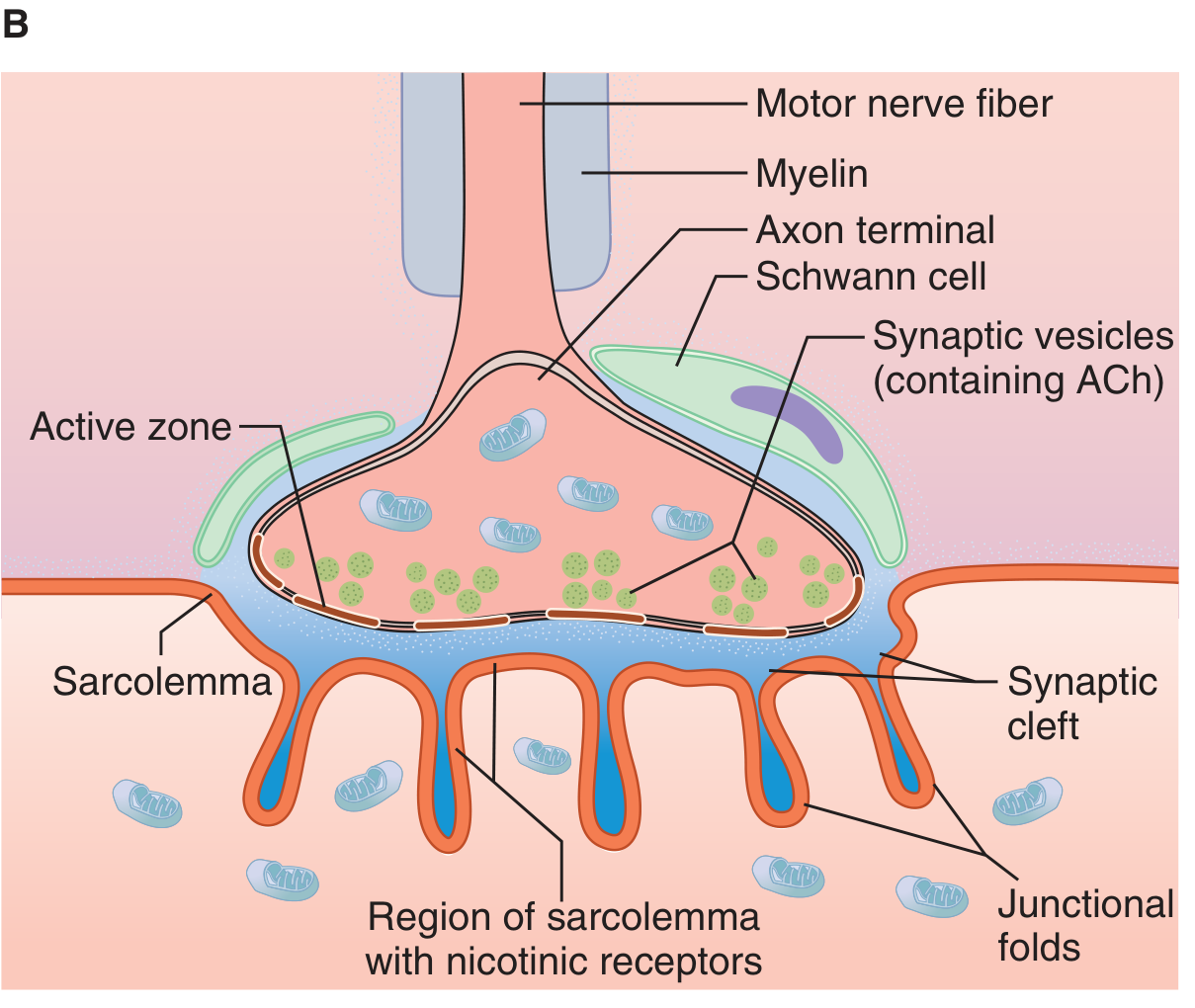
Figure: Neuromuscular junction structure - Ganong's Review of Medical Physiology, 26th Ed.
| Component | Description |
|---|---|
| Motor nerve terminal | Loses myelin sheath; divides into terminal boutons (axon terminals) |
| Axon terminal (presynaptic) | Contains mitochondria + synaptic vesicles loaded with ACh; has an "active zone" for vesicle docking |
| Synaptic cleft | ~50 nm gap between nerve and muscle; contains acetylcholinesterase (AChE) |
| Motor end plate (postsynaptic) | Thickened portion of the sarcolemma with junctional folds; rich in nicotinic ACh receptors (N_M type) at density ~10,000/µm² |
Each end plate receives input from a single nerve fiber. The whole structure -- nerve terminal + synaptic cleft + motor end plate -- is the NMJ.
2. Sequence of Neuromuscular Transmission
The steps follow an orderly cascade:
1. Action potential arrives at the motor nerve terminal
2. Voltage-gated Ca²+ channels open → Ca²+ influx into the presynaptic terminal
3. Vesicle exocytosis -- Ca²+ triggers fusion of ~60 ACh-containing vesicles with the presynaptic membrane. Each vesicle contains ~10,000 ACh molecules (quantal release).
4. ACh diffuses across the synaptic cleft to nicotinic N_M receptors on the junctional folds
5. Receptor activation -- Two ACh molecules bind to the α-β and δ-α subunit interfaces of the pentameric receptor (2α, β, δ, γ subunits). This opens the cation channel.
6. End plate potential (EPP) -- Na⁺ influx (and K⁺ efflux) generates a graded depolarization at the end plate. If the EPP is large enough, adjacent muscle membrane reaches threshold.
7. Action potential propagates bidirectionally along the entire muscle fiber → excitation-contraction coupling → muscle contracts.
8. ACh removal -- AChE in the synaptic cleft rapidly hydrolyzes ACh → choline + acetate. Choline is recycled back into the nerve terminal.
Quantal Release: At rest, random spontaneous vesicle fusion produces tiny "miniature end plate potentials" (MEPPs) ~0.5 mV each. Ca²+ increases quantal release; Mg²+ opposes it.
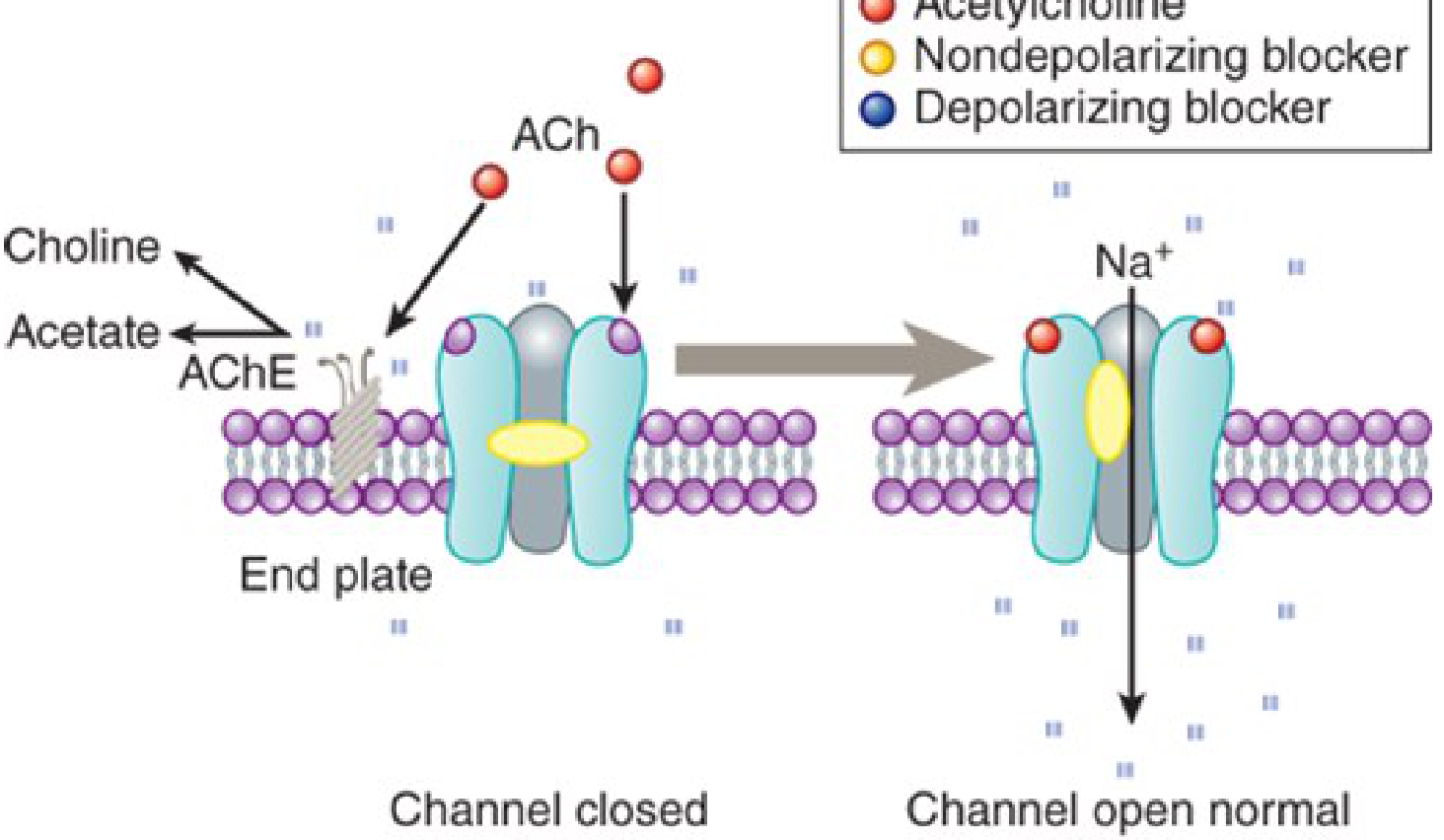
Figure: End plate channel interactions -- nondepolarizing (yellow) prevents opening; depolarizing (blue) blocks and persistently occupies the channel - Katzung's Basic & Clinical Pharmacology, 16th Ed.
3. The Nicotinic ACh Receptor (N_M)
- Pentameric structure: 2α + β + δ + γ (fetal) or ε (adult) subunits; each has 4 transmembrane domains (M1-M4); M2 lines the channel pore
- Binding of two ACh molecules (one at α-β interface, one at δ-α interface) is required for channel opening
- The receptor is a ligand-gated ion channel - it does not require G-proteins
- Two additional receptor types exist within the NMJ apparatus:
- Presynaptic receptors on the motor axon terminal: activation mobilizes more ACh vesicles toward the membrane for release
- Extrajunctional receptors: normally sparse but proliferate with prolonged immobilization or burns (clinically relevant for succinylcholine - risk of life-threatening hyperkalemia)
4. Neuromuscular Blocking Drugs
All NM blockers: structurally resemble ACh, contain quaternary nitrogen(s) (poorly lipid soluble, cannot cross CNS), are inactive orally, must be given parenterally.
Blockade occurs by two fundamentally different mechanisms:
A. Nondepolarizing (Competitive) Blockers
Prototype: d-Tubocurarine (curare) - the original from Amazonian arrow poison
Mechanism: Competitive antagonists at the nicotinic ACh receptor - bind to α subunits and prevent ACh from opening the channel. They do NOT depolarize the end plate.
Key features:
- Produce train-of-four (TOF) fade on repetitive nerve stimulation
- Reversed by acetylcholinesterase inhibitors (neostigmine, pyridostigmine, edrophonium) + atropine, or by sugammadex (for rocuronium/vecuronium)
- Fade in TOF signifies blockade
Drugs and classification:
| Family | Drugs | Duration | Elimination |
|---|---|---|---|
| Isoquinoline | d-Tubocurarine, Atracurium, Cisatracurium, Mivacurium | Variable | Hofmann elimination (atracurium - organ-independent), renal/plasma |
| Steroid | Pancuronium, Vecuronium, Rocuronium, Pipecuronium | Varies | Hepatic (3-hydroxy metabolites ~40-80% potency); renal for pancuronium |
- Short-acting: Mivacurium (~15 min), Rocuronium (intermediate, ~30-60 min)
- Long-acting: Pancuronium (>35 min, renal excretion)
- Steroidal agents are metabolized in the liver to 3-hydroxy/17-hydroxy metabolites (still have partial activity)
B. Depolarizing Blockers
Only clinically used drug: Succinylcholine (suxamethonium) -- structurally it is two ACh molecules linked end-to-end
Mechanism: Acts as a nicotinic agonist -- binds the receptor, opens the channel, depolarizes the end plate, then remains bound (because it is resistant to AChE). Sustained depolarization prevents re-firing.
Phase I block: Initial fasciculations followed by flaccid paralysis; all four TOF twitches are equally reduced (no fade)
Phase II block (desensitization block): With repeated or large doses, the receptor desensitizes. TOF shows fade, resembling a nondepolarizing block.
Pharmacokinetics: Extremely short duration (5-10 min) - rapidly hydrolyzed by plasma pseudocholinesterase (butyrylcholinesterase). Only a small fraction reaches the NMJ. Prolonged blockade occurs in:
- Genetic variants of plasma cholinesterase (detected by dibucaine number - normal enzyme inhibited 80%, abnormal only 20%)
- Liver disease, pregnancy, organophosphate poisoning
Cautions/contraindications:
- Burns, crush injuries, prolonged immobilization, denervation → proliferation of extrajunctional receptors → succinylcholine causes massive K⁺ release → life-threatening hyperkalemia
- Malignant hyperthermia (with volatile anesthetics)
- Not reversible by anticholinesterases (makes it worse - increases ACh, worsens depolarization)
5. Monitoring Neuromuscular Blockade
Train-of-Four (TOF): Four stimuli at 2 Hz (0.5 sec intervals)
- Depolarizing block: all four twitches equally reduced
- Nondepolarizing block: progressive fade (4th twitch weaker than 1st); TOF ratio <0.7 = inadequate recovery
- Full clinical recovery requires TOF ratio >0.9
Tetanic stimulation (30-100 Hz): Nondepolarizing block shows fade + posttetanic facilitation
6. Reversal of Neuromuscular Blockade
| Agent | Mechanism | Used For |
|---|---|---|
| Neostigmine | AChE inhibitor (+ atropine to block muscarinic side effects) | Nondepolarizing blockers |
| Pyridostigmine | AChE inhibitor | Nondepolarizing blockers; also used in myasthenia gravis |
| Edrophonium | Short-acting AChE inhibitor | Diagnosis (Tensilon test) + reversal |
| Sugammadex | Encapsulates rocuronium/vecuronium (cyclodextrin) | Rocuronium, vecuronium (even deep block) |
7. Toxins Affecting the NMJ
Botulinum Toxin (Clostridium botulinum)
- A family of 7 neurotoxins (A-G); A, B, E most toxic to humans
- Mechanism - PRESYNAPTIC: Cleaves SNARE proteins → prevents ACh vesicle fusion and release
- Toxin A and E: cleave SNAP-25 (synaptosome-associated protein 25 - needed for vesicle fusion with presynaptic membrane)
- Toxin B: cleaves synaptobrevin/VAMP (vesicle-associated membrane protein)
- Result: flaccid paralysis (decreased ACh release)
- EMG: facilitating pattern on repetitive stimulation (amplitude increases - postsynaptic receptors intact)
- Clinical: ptosis, diplopia, dysarthria, dysphagia, respiratory failure; also autonomic features (dry mouth, blurred vision)
- Therapeutic use (botox): local injection for achalasia, wrinkles, spasticity, dystonia, hyperhidrosis, migraine
Tetanus Toxin (Clostridium tetani)
- Binds presynaptic membrane at NMJ → travels retrograde via axonal transport to spinal cord motor neuron → taken up by inhibitory interneuron terminals
- Cleaves synaptobrevin (like BoTox B) but in glycinergic/GABAergic terminals
- Blocks release of inhibitory neurotransmitters (glycine, GABA) → disinhibition of motor neurons → spastic paralysis ("lockjaw"/trismus, opisthotonus)
α-Bungarotoxin (Bungarus, Cobra venoms)
- α-toxins (~7 kDa peptides) from krait (Bungarus multicinctus) and cobra (Naja naja)
- POSTSYNAPTIC block: Bind irreversibly to nicotinic ACh receptors
- Historically used as probes to isolate and characterize nicotinic receptors
Organophosphates and Nerve Agents
- Irreversible AChE inhibitors (e.g., sarin, VX, parathion)
- ACh accumulates massively in the synaptic cleft → sustained depolarizing block (Phase II-like) + cholinergic crisis (muscarinic + nicotinic effects)
- Treatment: atropine (muscarinic block) + pralidoxime (reactivates AChE if given early, before "aging")
Tick Paralysis
- Tick saliva neurotoxin → blocks ACh release from presynaptic terminal
- Ascending flaccid paralysis; resolves with tick removal
Black Widow Spider Venom (α-Latrotoxin)
- Causes massive, uncontrolled exocytosis of ACh vesicles → initial paradoxical excessive release, then depletion
- Painful muscle cramps, spasms
Hypermagnesemia
- Mg²+ competes with Ca²+ at the presynaptic terminal → reduces Ca²+-dependent ACh release
- Seen in: toxemia of pregnancy treated with parenteral MgSO₄, Mg-containing antacids
- Causes progressive weakness, respiratory failure
Hypophosphatemia
- Impairs ATP production → impairs vesicle recycling and transmitter synthesis
- Seen in: total parenteral nutrition, phosphate-binding antacids, severe respiratory alkalosis
8. Diseases of the Neuromuscular Junction
Myasthenia Gravis (MG)
Most common NMJ disease. Autoimmune.
Pathophysiology:
- ~85% of patients: Anti-AChR antibodies (IgG) → complement-mediated damage + receptor internalization/degradation → fewer functional receptors on the end plate
- ~10-15%: Anti-MuSK antibodies (muscle-specific tyrosine kinase) → impairs clustering of ACh receptors at the end plate
- Result: smaller EPPs → threshold not reached with fatigue → fatigable weakness
Clinical features:
- Ocular muscles almost always affected: ptosis, diplopia (these muscles are most sensitive as their motor units have highest safety factor demand)
- Bulbar: dysphagia, dysarthria, dysphonia
- Limb weakness: proximal > distal
- Symptoms worsen with repetitive use (fatigability), improve with rest
- Myasthenic crisis: respiratory failure (medical emergency)
Associations:
- Thymic hyperplasia (~65% of patients with AChR-MG)
- Thymoma (~15%) - always rule out
Diagnosis:
- EMG: decremental response to repetitive 3 Hz nerve stimulation (evoked compound muscle action potential amplitude falls >10%)
- Tensilon (edrophonium) test: IV edrophonium → rapid, dramatic improvement (AChE inhibitor reverses weakness briefly)
- Serology: anti-AChR antibodies; if negative, test for anti-MuSK
Treatment:
- AChE inhibitors: pyridostigmine (first line for symptomatic relief)
- Immunosuppression: prednisone, azathioprine, mycophenolate
- Thymectomy (especially if thymoma, or generalized MG age <60)
- Crisis: plasma exchange (PLEX) or IV immunoglobulin (IVIG), mechanical ventilation
Lambert-Eaton Myasthenic Syndrome (LEMS)
Autoimmune, paraneoplastic in most cases.
Pathophysiology:
- Anti-VGCC antibodies (voltage-gated calcium channel, presynaptic P/Q type) → impaired Ca²+ entry → reduced quantal ACh release
- Most common malignancy: small cell lung cancer (~60%)
EMG: facilitating pattern - on repetitive stimulation, amplitude increases (opposite of MG); posttetanic facilitation prominent; first stimulus is weak but subsequent ones potentiate.
Clinical features:
- Proximal limb weakness (legs > arms); less ocular involvement than MG
- Autonomic dysfunction (dry mouth, impotence, constipation) - very characteristic
- Reflexes absent but may return after brief exercise (postexercise facilitation)
Treatment:
- 3,4-diaminopyridine (amifampridine): blocks presynaptic K+ channels → prolongs action potential → more Ca²+ entry → more ACh release
- Treat underlying malignancy
- Immunosuppression (IVIG, plasma exchange, steroids)
Congenital Myasthenic Syndromes (CMS)
Not autoimmune - caused by genetic mutations in NMJ proteins. Three groups:
| Type | Defect | Example | Treatment |
|---|---|---|---|
| Presynaptic | Choline acetyltransferase absent/reduced → impaired ACh synthesis; or decreased quantal release | ChAT mutations | AChE inhibitors |
| Synaptic cleft | AChE deficiency → prolonged EPP, repetitive firing, end plate myopathy | Collagen Q tail mutations | Avoid AChE inhibitors (worsens); quinidine |
| Postsynaptic - Slow channel | Gain-of-function mutations in AChR subunits → prolonged channel opening → excessive stimulation, end plate myopathy | AChR subunit mutations | Quinidine, fluoxetine (channel blockers); avoid AChE inhibitors |
| Postsynaptic - Fast channel | Loss-of-function mutations → rapid channel closing → brief EPP | AChR subunit mutations | AChE inhibitors + 3,4-diaminopyridine |
Common features of all CMS: positive family history, fatigable weakness since infancy, ptosis, decremental EMG, negative anti-AChR antibodies.
Botulism (also a toxin-mediated NMJ disease)
- Presynaptic block of ACh release (see toxins section)
- EMG: facilitating pattern (like LEMS) - increment on rapid stimulation
- Treatment: antitoxin, supportive (ventilator if respiratory failure)
Organophosphate/Nerve Agent Toxicity
- Cholinergic crisis from AChE inhibition
- Classic SLUDGE/DUMBELS syndrome (excessive muscarinic effects) + nicotinic effects (muscle fasciculations → paralysis)
- Treatment: atropine + pralidoxime (before "aging" of enzyme)
9. Summary Comparison Table: Key NMJ Diseases
| Feature | Myasthenia Gravis | Lambert-Eaton (LEMS) | Botulism |
|---|---|---|---|
| Site | Postsynaptic | Presynaptic | Presynaptic |
| Antibody/Toxin | Anti-AChR, anti-MuSK | Anti-VGCC | BoNT cleaves SNAREs |
| Weakness distribution | Ocular/bulbar prominent | Proximal limbs prominent | Descending, cranial first |
| Autonomic features | Absent | Prominent (dry mouth) | Prominent (pupil dilation, dry mouth) |
| EMG repetitive stimulation | Decrement (low frequency) | Increment (high frequency) | Increment (like LEMS) |
| Reflexes | Normal | Absent (return post-exercise) | Decreased/absent |
| Treatment | Pyridostigmine, immunosuppression, thymectomy | 3,4-DAP, treat cancer, immunosuppression | Antitoxin, ventilatory support |
Sources
- Ganong's Review of Medical Physiology, 26th Ed., pp. 141-142
- Katzung's Basic and Clinical Pharmacology, 16th Ed., pp. 743-756
- Goodman & Gilman's Pharmacological Basis of Therapeutics, pp. 387-400
- Eric Kandel - Principles of Neural Science, 6th Ed., pp. 1478-1490
- Robbins & Kumar Basic Pathology, pp. 717-718
- Bradley and Daroff's Neurology in Clinical Practice, 6th Ed.
- Goldman-Cecil Medicine, Chapter 390
This is a shared conversation. Sign in to Orris to start your own chat.