Acute lymphoblastic leukemia
Reading File
search_medical_text
acute lymphoblastic leukemia pathophysiology classification diagnosis
search_medical_text
acute lymphoblastic leukemia treatment prognosis risk stratification
search_medical_image
acute lymphoblastic leukemia bone marrow peripheral blood smear
GradeResponse
GradeResponse
GradeResponse
Acute Lymphoblastic Leukemia (ALL)
Overview
ALL is a malignant clonal proliferation of lymphoid progenitor cells (lymphoblasts) that accumulate in the bone marrow, peripheral blood, and extramedullary sites. It is the most common childhood cancer (~80% of pediatric leukemias) but also occurs in adults, where prognosis is considerably worse.
Classification
ALL is classified by lineage and cytogenetics/molecular features (Harrison's, p. 3175):
| Category | Subtype | Key Features |
|---|---|---|
| B-cell lineage | B-ALL | ~85% of cases; CD19+, CD10+/- |
| T-cell lineage | T-ALL | ~15%; CD3+, CD7+; mediastinal mass common |
Key Cytogenetic/Molecular Subtypes
| Subtype | Frequency | Prognosis |
|---|---|---|
| t(12;21) ETV6-RUNX1 | ~25% pediatric | Favorable |
| Hyperdiploidy (>50 chromosomes) | ~25% pediatric | Favorable |
| t(9;22) BCR-ABL1 (Ph+) | ~25–30% adults | Poor (improved with TKIs) |
| Ph-like ALL | ~15–20% adults | Poor (targetable kinase lesions) |
| t(4;11) MLL-AF4 (KMT2A) | Infants | Poor |
| Hypodiploidy (<44 chromosomes) | ~1–2% | Poor |
| iAMP21 | ~2% pediatric | Intermediate-poor |
Ph-like (BCR-ABL1–like) ALL carries a gene expression profile similar to Ph+ ALL without the BCR-ABL1 fusion, harbors targetable kinase lesions (JAK2, CRLF2, ABL-class fusions), and is associated with minimal residual disease (MRD) persistence and poor outcomes (Management of Adult ALL, p. 25).
Pathophysiology
Lymphoblasts arise from committed lymphoid progenitors due to acquired genetic alterations:
- Chromosomal translocations creating fusion oncoproteins (e.g., BCR-ABL1, ETV6-RUNX1)
- Copy number alterations (e.g., IKZF1/Ikaros deletion — independently associated with poor prognosis)
- Gene expression dysregulation driving uncontrolled proliferation and block in differentiation
- Blast accumulation suppresses normal hematopoiesis → cytopenias
Clinical Presentation
Symptoms of bone marrow failure:
- Fatigue, pallor (anemia)
- Fever, infections (neutropenia)
- Bleeding, petechiae, bruising (thrombocytopenia)
- Bone pain (especially in children)
Extramedullary involvement:
- Hepatosplenomegaly, lymphadenopathy
- CNS involvement — headache, cranial nerve palsies, meningism (more common in T-ALL and high-risk B-ALL)
- Anterior mediastinal mass — classic for T-ALL; may cause SVC syndrome or respiratory compromise
Diagnosis
Per Harrison's (p. 3175), diagnosis requires:
- Peripheral blood smear — circulating blasts (variable; may be absent in aleukemic presentations)
- Bone marrow aspirate/biopsy — ≥20% lymphoblasts (WHO criteria); typically hypercellular with monotonous lymphoblast infiltrate
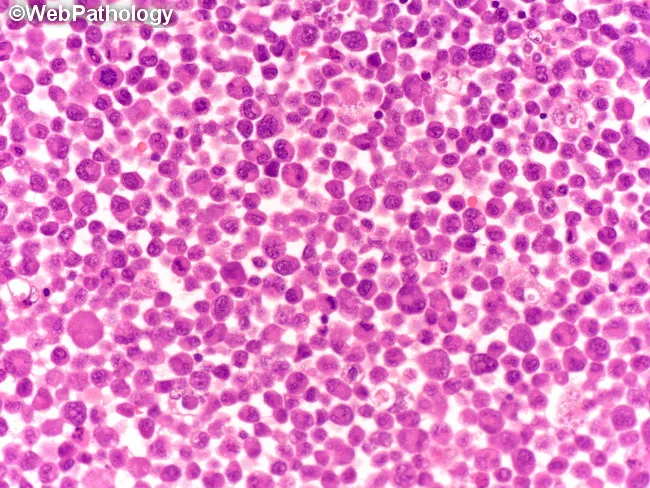
- Immunophenotyping (flow cytometry) — essential for B vs. T lineage assignment; key markers:
- B-ALL: CD19, CD10, CD22, CD79a, TdT
- T-ALL: CD3 (cytoplasmic), CD7, CD2, CD5, TdT
- Cytogenetics (karyotype + FISH) — identifies prognostic translocations
- Molecular studies — BCR-ABL1 PCR, Ph-like gene expression panel, IKZF1 deletion
- CSF analysis — lumbar puncture for CNS staging (CNS1/2/3)
- CBC, LFTs, uric acid, LDH — baseline labs; high LDH reflects tumor burden
Risk Stratification
Pediatric (COG risk groups)
| Group | Criteria |
|---|---|
| Standard risk | Age 1–9.99 yrs, WBC <50,000/µL, B-ALL, favorable cytogenetics |
| High risk | Age ≥10 or WBC ≥50,000/µL |
| Very high risk | Ph+, Ph-like, hypodiploidy, MLL rearrangement, poor MRD response |
Adults
Risk factors for poor prognosis include:
- Age >35–60 (worsens progressively)
- WBC >30,000 (B-ALL) or >100,000 (T-ALL)
- Ph+ or Ph-like genotype
- IKZF1 deletion
- Persistent MRD after induction
Treatment
General Framework
Treatment is delivered in 3 phases:
1. Induction (4–8 weeks)
Goal: achieve complete remission (CR) — <5% blasts in marrow
Pediatric regimens (e.g., AALL0434, DFCI):
- Vincristine + corticosteroid (dexamethasone preferred) + asparaginase ± anthracycline
Adult regimens (e.g., HyperCVAD, BFM-inspired):
- Vincristine, daunorubicin/doxorubicin, cyclophosphamide, corticosteroids, asparaginase
- CR rates: ~85–95% in pediatric, ~80–90% in adults
Ph+ ALL: Add a TKI (imatinib, dasatinib, or ponatinib) to chemotherapy backbone from day 1.
2. Consolidation / Intensification
- Multiple cycles of high-dose methotrexate (HD-MTX), cytarabine, 6-mercaptopurine
- CNS prophylaxis: intrathecal MTX ± cytarabine ± hydrocortisone (cranial irradiation largely abandoned)
- Allogeneic SCT in high-risk adults achieving CR1 (Ph+, MRD+, adverse cytogenetics)
3. Maintenance (~2–3 years)
- Daily 6-mercaptopurine + weekly methotrexate ± monthly vincristine/corticosteroid pulses
- MRD monitoring guides intensification
Targeted / Novel Agents
| Agent | Indication |
|---|---|
| Imatinib / Dasatinib / Ponatinib | Ph+ ALL (TKIs) |
| Blinatumomab (CD19×CD3 BiTE) | Relapsed/refractory B-ALL; MRD+ disease |
| Inotuzumab ozogamicin | Relapsed/refractory CD22+ B-ALL |
| Tisagenlecleucel (CAR-T) | Pediatric/young adult r/r B-ALL |
| Nelarabine | T-ALL (nucleoside analog) |
| Venetoclax | Emerging role, especially in elderly |
Blinatumomab + ponatinib combinations have shown activity in relapsed/refractory Ph+ ALL (Management of Adult ALL, p. 25).
CNS Disease
- Prophylaxis is mandatory for all patients (intrathecal chemotherapy)
- CNS3 (blasts in CSF + symptoms) requires intensified IT therapy ± cranial radiation
- CNS relapse carries poor prognosis
Complications
| Complication | Cause |
|---|---|
| Tumor lysis syndrome | Rapid cell death at induction |
| Infections | Prolonged neutropenia, immunosuppression |
| Osteonecrosis | Corticosteroids (especially adolescents) |
| Pancreatitis / hepatotoxicity | Asparaginase |
| Cardiotoxicity | Anthracyclines |
| Neuropathy | Vincristine |
| Secondary malignancy | Late effect of therapy |
Prognosis
| Population | 5-Year OS |
|---|---|
| Pediatric (standard risk) | ~90–95% |
| Pediatric (high risk) | ~75–85% |
| Adults <40 years | ~40–60% |
| Adults >60 years | ~20–30% |
| Ph+ ALL (TKI era) | ~50–60% (improving) |
| Relapsed/refractory | ~30–40% (with novel agents) |
Minimal residual disease (MRD) — detected by flow cytometry or PCR — is the single most important prognostic marker post-induction. MRD negativity predicts sustained remission; MRD positivity should prompt treatment intensification.
Special Populations
- Infants (<1 year): Often KMT2A-rearranged; very poor prognosis; experimental protocols
- Adolescents & young adults (AYA): Pediatric-inspired regimens (with asparaginase) outperform adult regimens
- Elderly (>65): Reduced-intensity regimens; blinatumomab-based approaches gaining ground
- Down syndrome: Hypersensitive to methotrexate; modified protocols required
This is a shared conversation. Sign in to Orris to start your own chat.