Biological carcinogenesis for 10 marker question
Biological Carcinogenesis
Definition
1. Oncogenic RNA Viruses (Retroviruses)
Human T-Cell Leukemia Virus Type 1 (HTLV-1)
- Tropism for CD4+ T cells
- Transmission via sexual intercourse, blood products, or breastfeeding
- Leukemia develops in only 3-5% of infected individuals after a latent period of 40-60 years
- HTLV-1 does not carry a recognizable oncogene and shows no consistent proto-oncogene insertion
- The HTLV-1 genome contains tax and HBZ genes beyond the standard gag/pol/env regions
- Tax protein: stimulates transcription of viral RNA from the 5' long terminal repeat; alters transcription of host cell genes; interacts with cell signaling proteins
- HBZ: a transcription factor that, along with Tax, contributes to genomic instability, inhibition of senescence, and altered growth signals
- The integration pattern is clonal in leukemic cells, confirming infection preceded transformation
2. Oncogenic DNA Viruses
A. Human Papillomavirus (HPV)
- Low-risk types (HPV-6, HPV-11): cause benign genital warts, low malignant potential
- High-risk types (HPV-16, HPV-18): cause squamous cell carcinomas of the cervix, anogenital region, and head & neck (especially tonsillar mucosa)
- In benign warts: HPV genome is episomal (non-integrated)
- In cancers: HPV genome is integrated into the host genome, always within the E1/E2 open reading frame - this destroys the E2 viral repressor and leads to overexpression of E6 and E7 oncoproteins
- Binds and degrades p53 (tumor suppressor)
- Stimulates expression of TERT (telomerase reverse transcriptase) - contributing to cellular immortalization
- E6 from high-risk types has higher affinity for p53 than E6 from low-risk types
- Binds RB protein and displaces E2F transcription factors, driving cells through the G1/S checkpoint
- Inactivates CDK inhibitors p21 and p27
- Activates cyclins A and E
- E7 from high-risk HPV types has higher affinity for RB
Clinical relevance: HPV vaccines have been proven effective in preventing cervical cancer, confirming HPV's primary role.
B. Epstein-Barr Virus (EBV)
- Burkitt lymphoma (particularly the endemic African form)
- Hodgkin lymphoma (mixed cellularity subtype)
- Nasopharyngeal carcinoma
- Post-transplant lymphoproliferative disorders / immunosuppression-related lymphomas
- EBV infects B lymphocytes via the CD21 receptor
- Expresses LMP-1 (Latent Membrane Protein-1) which mimics constitutively active CD40 signaling, activating NF-κB and promoting B cell survival and proliferation
- Also expresses EBNA-2, which transactivates cellular genes including cyclin D and c-myc
- In immunocompetent hosts, proliferating B cells are kept in check by T cells; in immunosuppressed individuals, polyclonal proliferation can progress to monoclonal lymphoma
C. Hepatitis B Virus (HBV) and Hepatitis C Virus (HCV)
- HBV DNA integrates into the host genome, causing chromosomal instability
- Chronic inflammation, hepatocyte necrosis, and regeneration drive accumulation of mutations
- HBV-encoded HBx protein transactivates many growth-promoting genes and impairs p53 function
- HCV is an RNA virus that does not integrate into DNA
- Carcinogenesis is driven primarily by chronic inflammation and cirrhosis, leading to ongoing cycles of cell death and regeneration, accumulating mutations over decades
3. Bacterial Carcinogenesis
Helicobacter pylori (H. pylori)
- Gastric adenocarcinoma
- Gastric MALT (mucosa-associated lymphoid tissue) lymphoma
- Triggers chronic active gastritis, leading to sustained epithelial proliferation
- CagA protein (encoded by cytotoxicity-associated gene) is injected into gastric epithelial cells via a type IV secretion system, where it activates signaling pathways that promote cell growth and inhibit apoptosis
- Chronic inflammation generates reactive oxygen species (ROS) that cause DNA damage
- MALT lymphoma develops from antigen-driven B cell proliferation; early-stage disease can regress with H. pylori eradication - confirming infection as the primary driver
4. Multistep Nature of Biological Carcinogenesis
- The long latent period between infection and malignancy
- The fact that only a minority of infected individuals develop cancer
- The need for additional co-carcinogens or immune suppression
Summary Table
| Agent | Cancer Type | Key Mechanism |
|---|---|---|
| HTLV-1 | Adult T-cell leukemia/lymphoma | Tax/HBZ proteins - altered T-cell signaling |
| HPV-16/18 | Cervical, anogenital, head & neck carcinoma | E6 degrades p53; E7 inactivates RB |
| EBV | Burkitt lymphoma, Hodgkin lymphoma, nasopharyngeal Ca | LMP-1 activates NF-κB; B cell immortalization |
| HBV | Hepatocellular carcinoma | HBx protein; chromosomal instability |
| HCV | Hepatocellular carcinoma | Chronic inflammation, cirrhosis |
| H. pylori | Gastric carcinoma, MALT lymphoma | CagA protein; chronic inflammation |
Can you make these answers in a flowchart pattern which are easy to remember
A detailed educational flowchart on Biological Carcinogenesis for medical students. Clean white background, organized top-down flowchart with color-coded boxes and arrows. Start at top with main title box: "BIOLOGICAL CARCINOGENESIS" (dark blue header) Branch into 3 main categories below it with arrows: 1. RNA VIRUS (orange box) → HTLV-1 → Infects CD4+ T cells → Tax + HBZ proteins → Altered signaling, genomic instability → Adult T-Cell Leukemia/Lymphoma (ATLL) [latency 40-60 yrs, only 3-5% develop cancer] 2. DNA VIRUSES (green box) → Three sub-branches: A. HPV (16,18) → Integration into host genome → Loss of E2 repressor → E6: degrades p53 + activates TERT / E7: inactivates RB, p21, p27, activates cyclins → Cervical, Anogenital, Head & Neck Ca B. EBV → CD21 receptor → LMP-1 mimics CD40 → NF-κB activation → B cell immortalization → Burkitt lymphoma, Hodgkin lymphoma, Nasopharyngeal Ca C. HBV/HCV → HBx protein (HBV) / Chronic inflammation (HCV) → Chromosomal instability + cirrhosis → Hepatocellular Carcinoma 3. BACTERIA (red box) → H. pylori → CagA protein injected into cells → NF-κB, ROS, DNA damage + Chronic inflammation → Gastric Adenocarcinoma + MALT Lymphoma At the bottom, convergence arrow into final box: "MULTISTEP CARCINOGENESIS: Multiple driver mutations accumulate over time → Full malignant transformation" Key concept note box on side: "Common theme: Infection → Polyclonal proliferation → Monoclonal cancer (with additional mutations)" Use distinct colors for each branch, bold labels, clean sans-serif font, medical textbook quality
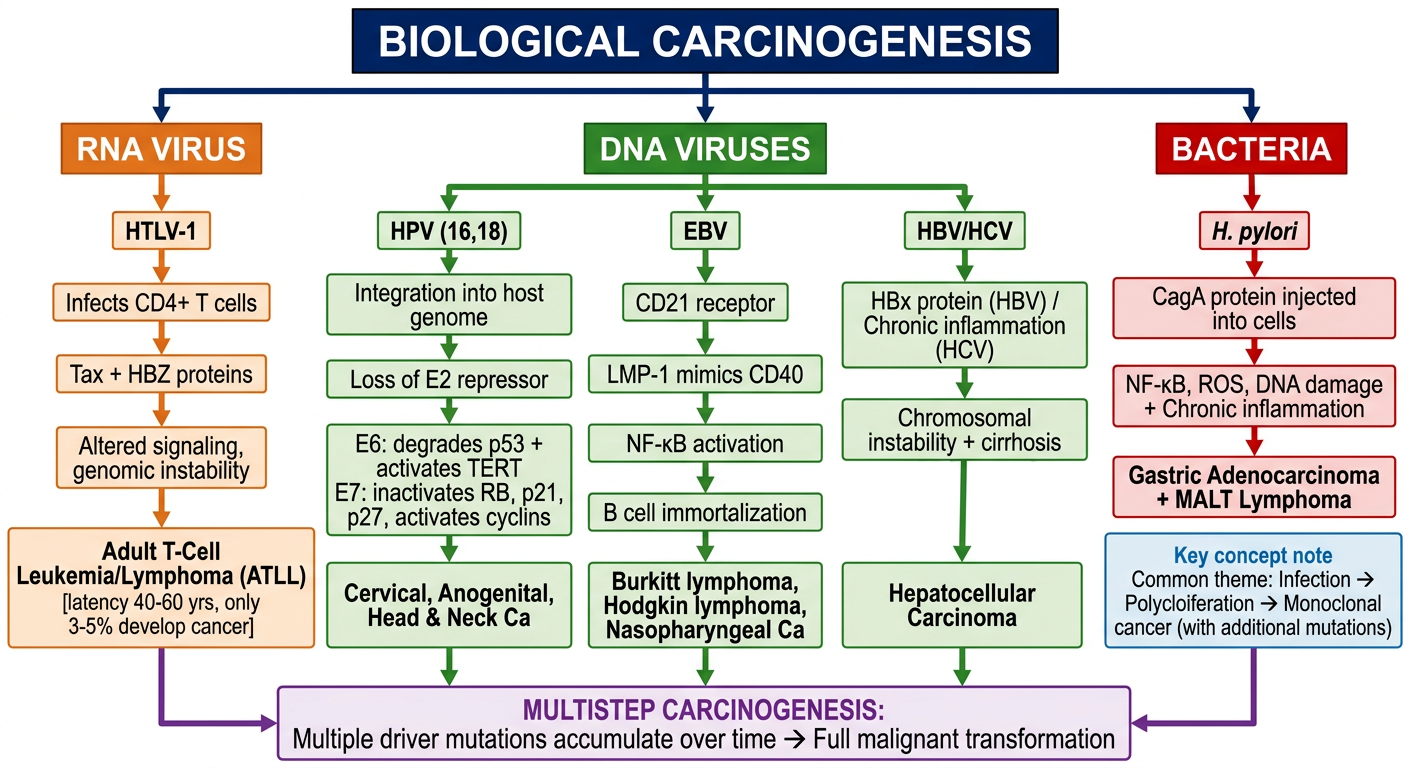
- HTLV-1 → T cells → Tax/HBZ → ATLL (think: "T-T-T" - HTLV, T cells, Tax)
- HPV 16/18 → E6 kills p53, E7 kills RB → Cervical Ca (E6=p53, E7=RB - alphabetical order)
- EBV → CD21 → LMP-1 → NF-κB → Burkitt/Hodgkin/Nasopharyngeal (3 cancers)
- HBV/HCV → Liver → HCC (Hepatitis = Hepatocellular)
- H. pylori → CagA → Gastric Ca + MALT (Cag = Cancer-associated gene)