How blood clot ?
Reading File
Finding Sources
Searching the Web
blood clotting coagulation cascade mechanism overview
Finding Sources
Finding Sources
Reading File
Reading File
Searching Images
blood coagulation cascade clotting factors diagram
Reading File
I now have excellent content from Robbins and Medical Physiology. Let me compile a comprehensive, well-organized answer.
How Blood Clots — The Mechanisms of Hemostasis
Blood clotting (hemostasis) is a tightly regulated response to vascular injury. It involves four overlapping steps: vascular spasm, platelet plug formation, coagulation cascade, and fibrinolysis.
Step 1 — Vascular Spasm
Immediately after injury, the damaged vessel constricts reflexively. This reduces blood flow and limits blood loss while the platelet and coagulation responses catch up. A tourniquet works on this same principle — increasing external pressure decreases the vessel's transmural pressure and flow.
Step 2 — Primary Hemostasis (Platelet Plug)
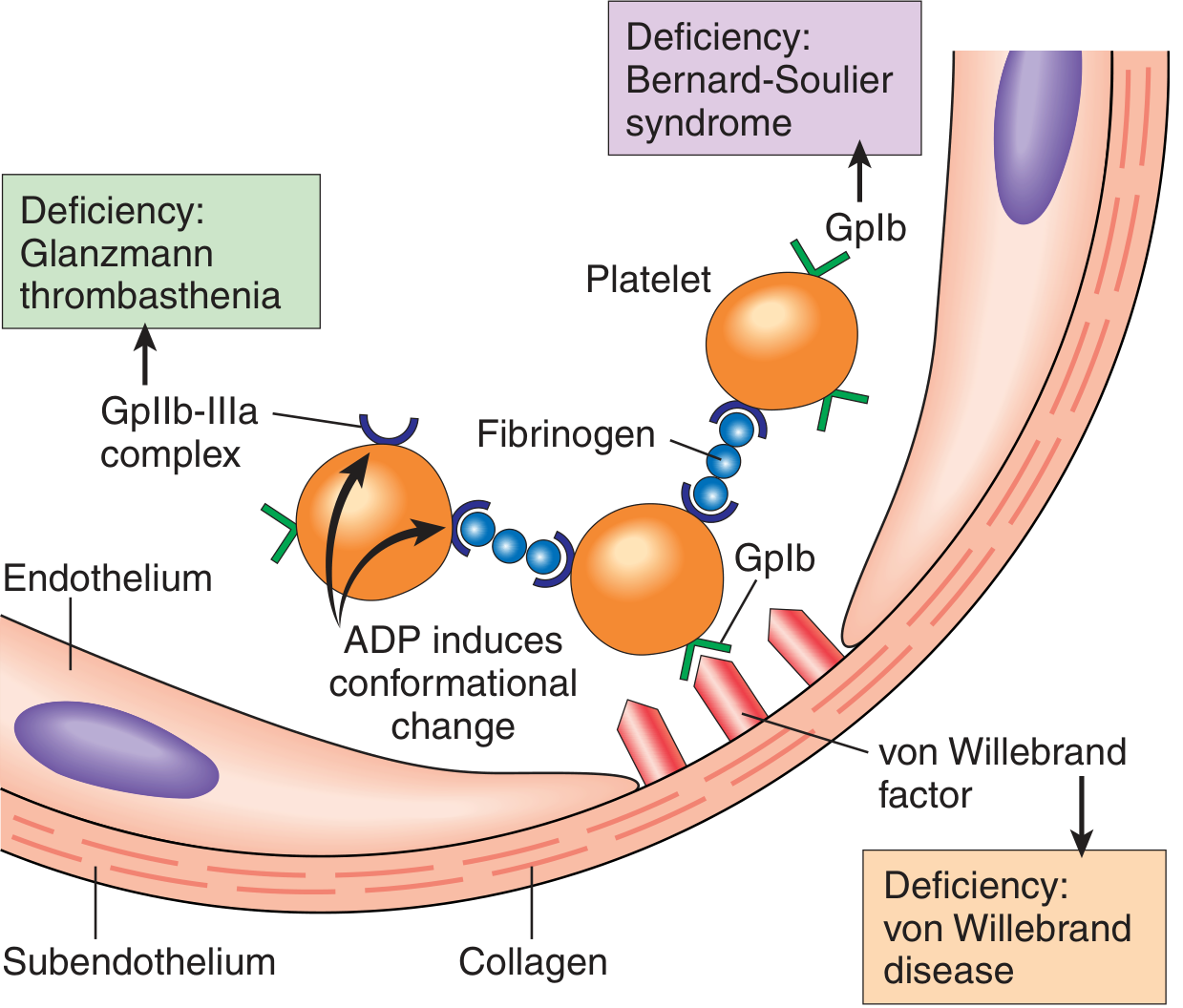
When the vessel wall is breached, the subendothelial matrix is exposed. Three sequential platelet events follow:
Adhesion
- Collagen and von Willebrand factor (vWF) — secreted by endothelial cells and stored in their Weibel-Palade bodies — are exposed.
- vWF acts as a molecular bridge, binding to glycoprotein Ib (GpIb) on the platelet surface.
- Collagen also binds directly to platelet GpIa/IIa. Genetic loss of vWF causes von Willebrand disease; loss of GpIb causes Bernard-Soulier syndrome, both presenting as bleeding disorders.
Activation
- Ligand binding triggers intracellular signaling → shape change (smooth disc → spiky "sea urchin" with greatly increased surface area).
- Two types of granules are released:
- α-granules: fibrinogen, factor V, vWF, fibronectin, PDGF, TGF-β, P-selectin
- Dense (δ) granules: ADP, ATP, calcium, serotonin, epinephrine
- ADP from dense granules recruits more platelets (recruitment amplification).
- Activated platelets produce thromboxane A₂ (TxA₂), a potent platelet aggregation inducer. Aspirin inhibits cyclooxygenase, blocking TxA₂ synthesis and impairing this step.
- Negatively charged phosphatidylserine flips to the outer leaflet, serving as a scaffold for coagulation factor assembly.
Aggregation
- Platelet activation changes the conformation of GpIIb/IIIa, allowing fibrinogen to bridge adjacent platelets → reversible platelet aggregate (primary plug).
- Loss of GpIIb/IIIa = Glanzmann thrombasthenia (bleeding disorder).
Step 3 — Secondary Hemostasis (Coagulation Cascade)
The coagulation cascade converts the soft, reversible platelet plug into a hard, stable fibrin clot. Each step involves an enzyme (activated factor) + substrate (proenzyme) + cofactor, all assembled on the phospholipid surface of activated platelets, in the presence of Ca²⁺.
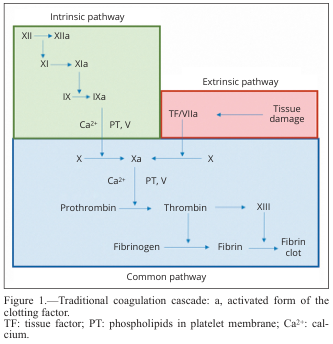
Two Initiation Pathways
| Pathway | Trigger | Lab Test |
|---|---|---|
| Extrinsic | Tissue factor (TF) released from injured tissue | Prothrombin Time (PT) |
| Intrinsic | Contact with negatively charged surfaces (collagen, glass) | aPTT |
Both converge at Factor X → the common pathway.
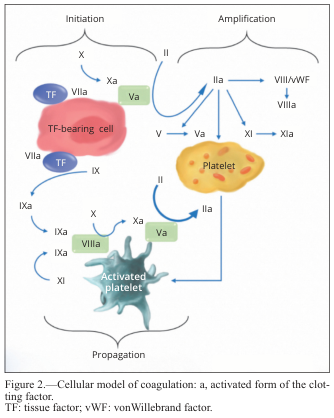
The Cascade in vivo
In living tissue, the extrinsic pathway dominates:
- Tissue factor (TF) is exposed at the injury site
- TF binds Factor VIIa → activates Factor IX and Factor X
- Factor Xa (with cofactor Va) forms the prothrombinase complex → converts prothrombin (II) → thrombin (IIa)
- Thrombin is the central enzyme — it:
- Cleaves fibrinogen → fibrin monomers
- Activates Factor XIII → crosslinks fibrin into a stable mesh
- Activates Factors V, VIII, XI (positive feedback amplification)
- Activates platelets via protease-activated receptors (PAR)
The intrinsic pathway (Factors XII → XI → IX → VIII) amplifies thrombin generation, especially via Factor XIa feedback — explaining why Factor XII deficiency causes no bleeding but Factor VIII/IX deficiency causes hemophilia.
Vitamin K Dependence
Factors II, VII, IX, X require vitamin K for γ-carboxylation of glutamic acid residues — this allows them to bind Ca²⁺ and assemble on phospholipid surfaces. Warfarin inhibits vitamin K recycling, blocking this process.
Step 4 — Anticoagulant Mechanisms (Limits to Clotting)
The body prevents clotting from spreading beyond the injury site:
| Inhibitor | Action |
|---|---|
| Antithrombin (AT-III) | Inactivates thrombin and Factor Xa; potentiated by heparin |
| Protein C/S | Thrombin activates Protein C (via thrombomodulin on endothelium); Protein C + Protein S degrade Factors Va and VIIIa |
| TFPI (Tissue Factor Pathway Inhibitor) | Inhibits the TF/VIIa complex |
| Prostacyclin (PGI₂) | Released by intact endothelium; inhibits platelet activation |
Step 5 — Fibrinolysis (Clot Dissolution)
Once the vessel heals, the clot is dissolved:
- Plasminogen (embedded in the clot) is activated to plasmin by tissue plasminogen activator (tPA) released from endothelial cells.
- Plasmin degrades the fibrin mesh → fibrin degradation products (FDPs/D-dimers).
- FDPs themselves inhibit thrombin, providing further anticoagulant feedback.
- PAI-1 (plasminogen activator inhibitor-1) limits excessive fibrinolysis.
Summary Flowchart
Vascular injury
↓
Vascular spasm + collagen/TF exposure
↓
Platelet adhesion (vWF–GpIb) → Activation → Aggregation (GpIIb/IIIa–fibrinogen)
↓ ↓
Primary platelet plug Phospholipid scaffold
↓
TF + VIIa → IX/X activation (Extrinsic)
XII→XI→IX→VIII (Intrinsic amplification)
↓
Prothrombinase complex (Xa + Va)
↓
Prothrombin → THROMBIN (IIa)
↓
Fibrinogen → Fibrin → Crosslinked fibrin (XIIIa)
↓
Stable secondary hemostatic plug
↓
Fibrinolysis (plasmin) → clot dissolution
Sources:
- Robbins & Kumar Basic Pathology, pp. 83–86
- Medical Physiology (Boron & Boulpaep), pp. 641–643
- Perioperative Drug and Hemostasis Management in Vascular Surgery (cascade diagrams)
This is a shared conversation. Sign in to Orris to start your own chat.