Hypertension= causes physiology, pathology, pharmacology , treatment, reason
Reading File
Finding Sources
Finding Sources
Searching PubMed
"hypertension" AND "pathophysiology"
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
I now have comprehensive information from multiple authoritative textbooks. Let me compile the full, detailed response.
Hypertension: Complete Overview
1. Definition & Classification
Blood pressure (BP) = Cardiac Output (CO) × Peripheral Vascular Resistance (PVR). Hypertension exists when BP is persistently elevated. Standard classification (AHA/ACC):
| Category | Systolic (mmHg) | Diastolic (mmHg) |
|---|---|---|
| Normal | <120 | <80 |
| Elevated | 120-129 | <80 |
| Stage 1 | 130-139 | 80-89 |
| Stage 2 | ≥140 | ≥90 |
| Hypertensive crisis | >180 | >120 |
2. Causes
A. Primary (Essential) Hypertension - 90-95% of all cases
The cause is multifactorial and often not fully identifiable. Key contributing factors:
- Obesity/overweight: The single largest contributor - accounts for 65-75% of the risk. Framingham data show ~78% of primary hypertension in men and 65% in women can be ascribed to excess weight gain. Visceral fat compresses kidneys, raises intra-abdominal pressure (up to 35-40 mmHg), activates RAAS, and drives sympathetic tone.
- High sodium intake: Impairs renal pressure natriuresis, increases fluid retention.
- Sedentary lifestyle
- Excess alcohol consumption
- Low potassium intake
- Genetic predisposition: Polygenic susceptibility; positive family history is a major risk factor.
- Age: Vascular stiffness increases with age, raising systolic BP.
- Race: African Americans have higher prevalence and severity.
Rare monogenic causes (<1% of hypertension):
Most involve increased renal tubular sodium reabsorption or excess mineralocorticoid activity:
- Liddle syndrome (gain-of-function ENaC mutation)
- Gordon syndrome (increased NaCl cotransporter activity)
- Familial hyperaldosteronism types I & II
- Apparent mineralocorticoid excess (AME)
- Congenital adrenal hyperplasia (DOC overproduction)
B. Secondary Hypertension - 5-10% of cases
Specific identifiable cause; must be considered especially in young patients or resistant hypertension:
| System | Cause |
|---|---|
| Renal | Renovascular disease (renal artery stenosis), CKD, polycystic kidney disease, glomerulonephritis |
| Endocrine | Primary hyperaldosteronism (Conn's syndrome), pheochromocytoma, Cushing's syndrome, hypothyroidism/hyperthyroidism, acromegaly |
| Vascular | Coarctation of the aorta |
| CNS | Raised intracranial pressure (Cushing's reflex) |
| Drugs/toxins | Oral contraceptives, NSAIDs, sympathomimetics, cocaine, amphetamines, cyclosporin, steroids, licorice |
| Sleep | Obstructive sleep apnea |
| Pregnancy | Pre-eclampsia |
3. Physiology of Blood Pressure Regulation
BP is controlled through two major effector systems:
A. Renin-Angiotensin-Aldosterone System (RAAS) - Acts over minutes to hours
- Low renal perfusion pressure / low tubular NaCl / sympathetic stimulation → renin released from juxtaglomerular cells
- Renin cleaves angiotensinogen → Angiotensin I
- ACE (lung) converts Ang I → Angiotensin II (Ang II)
- Ang II effects:
- Direct vasoconstriction (AT1 receptors on vascular smooth muscle) → increases PVR
- Stimulates aldosterone release from adrenal cortex → Na+ and water retention → increases blood volume → increases CO
- Promotes renal tubular Na+ reabsorption directly
- Stimulates sympathetic nervous system
- Promotes vascular and cardiac hypertrophy (structural remodeling)
B. Sympathetic Nervous System (SNS) - Acts over seconds to minutes
- Norepinephrine binds α1-adrenoceptors on vessels → vasoconstriction → increased PVR
- Epinephrine binds β1-receptors on heart → increased heart rate and stroke volume → increased CO
- SNS stimulates renin release (β1 in kidney)
- SNS increases renal tubular Na+ reabsorption via direct innervation of nephrons
- Chronic increased sympathetic tone is a driver of sustained hypertension
C. Counter-regulatory Systems (that normally oppose hypertension)
- Kallikrein-kinin system: produces vasodilator kinins, stimulates prostaglandin and NO production
- Nitric oxide (NO): endothelium-derived vasodilator; deficiency promotes hypertension
- Natriuretic peptides (ANP, BNP): vasodilation + natriuresis; inhibit RAAS and SNS
- Endothelins: vasoconstrictors from endothelium that can contribute to hypertension
- Prostaglandin E & prostacyclin: counter Ang II and norepinephrine-mediated vasoconstriction
D. Renal Pressure Natriuresis
The kidney is the ultimate long-term controller of BP. Any rise in BP should trigger natriuresis (salt excretion) that reduces blood volume and returns BP to normal. Impairment of this mechanism - by renal disease, excess RAAS activity, or SNS activation - is a central defect in sustained hypertension.
4. Pathology (Structural Changes from Hypertension)
Hypertension causes end-organ damage through two main mechanisms: increased mechanical stress on vessel walls and neurohormonal activation (RAAS, SNS).
A. Vascular Pathology
- Hypertensive arteriosclerosis: medial hypertrophy of small arteries and arterioles, intimal thickening, and luminal narrowing
- Hyaline arteriolosclerosis: homogeneous pink thickening of arteriolar walls (protein deposition); seen in kidneys and retina
- Fibrinoid necrosis: in malignant hypertension - acute inflammatory necrosis of arteriolar walls, leads to thrombosis and ischemia
- Accelerated atherosclerosis: chronic hypertension damages endothelium, promotes lipid deposition and plaque formation in medium/large arteries
B. Cardiac Pathology (Hypertensive Heart Disease)
- Left ventricular hypertrophy (LVH): compensatory response to increased afterload; initially concentric (wall thickening, normal/small cavity), later eccentric (dilation)
- Diastolic dysfunction: stiff, hypertrophied ventricle - leads to heart failure with preserved ejection fraction (HFpEF)
- Systolic dysfunction: with prolonged hypertension, leads to heart failure with reduced EF (HFrEF)
- Coronary artery disease: accelerated by hypertension-driven atherosclerosis
- Risk: 2x increase in MI, stroke, HF, and sudden death
C. Renal Pathology (Hypertensive Nephropathy)
- Nephrosclerosis: hyalinization of afferent arterioles, glomerulosclerosis, tubular atrophy, interstitial fibrosis
- Glomerulomegaly and focal segmental glomerulosclerosis (FSGS): especially in obesity-related hypertension
- Proteinuria (can reach nephrotic range) followed by progressive CKD
- End-stage renal disease: hypertension + diabetes account for 70-75% of ESRD cases
D. Cerebrovascular Pathology
- Lacunar infarcts: small penetrating arteries undergo lipohyalinosis and occlusion
- Intracerebral hemorrhage: fibrinoid necrosis or Charcot-Bouchard microaneurysm rupture
- Hypertensive encephalopathy: failure of cerebral autoregulation at very high BP → cerebral edema
- Ischemic stroke: from accelerated atherosclerosis + thromboembolism
E. Retinal Pathology (Keith-Wagener-Barker Classification)
- Grade I: Mild arteriolar narrowing
- Grade II: A-V nicking (arteriovenous crossing changes)
- Grade III: Flame hemorrhages, cotton-wool spots
- Grade IV: Papilledema (indicates malignant hypertension)
F. Malignant Hypertension
A hypertensive emergency - BP typically >180/120 with end-organ damage. Characterized by:
- Progressive arteriopathy with fibrinoid necrosis of arterioles
- Renal involvement → renin release → further Ang II and aldosterone surge → vicious cycle
- Hypertensive encephalopathy, renal failure, cardiac failure, retinal changes (Grade III/IV)
5. Pharmacology & Treatment
Step 1: Non-Pharmacological (Lifestyle Modifications - first-line for Stage 1)
- Weight reduction (most effective single intervention)
- DASH diet (high fruits, vegetables, low saturated fat, reduced sodium)
- Sodium restriction (<2.3 g/day)
- Regular aerobic exercise (150 min/week moderate intensity)
- Alcohol limitation
- Smoking cessation (reduces overall cardiovascular risk)
Step 2: Pharmacological Treatment
A. Diuretics
| Drug | Class | Mechanism | Dose range | Key uses |
|---|---|---|---|---|
| Hydrochlorothiazide | Thiazide | Inhibit NaCl cotransporter (DCT) → reduce Na+/water reabsorption | 12.5-50 mg/day | First-line; all stages |
| Chlorthalidone | Thiazide-like | Same as above, longer half-life | 12.5-25 mg/day | Preferred over HCTZ in trials |
| Furosemide | Loop | Inhibit NKCC2 cotransporter (Loop of Henle) | 20-80 mg/day | CKD with reduced GFR, heart failure |
| Spironolactone | Aldosterone antagonist | Blocks mineralocorticoid receptor → inhibits Na+/K+-ATPase | 25-100 mg/day | Resistant hypertension, primary hyperaldosteronism |
| Amiloride | K+-sparing | Blocks ENaC in collecting duct | 5-10 mg/day | Liddle syndrome, adjunct |
Why they work: Reduce plasma volume → initially decrease CO; long-term, reduce PVR through vascular remodeling.
B. Beta-Adrenoceptor Blockers (β-blockers)
| Drug | Selectivity | Notes |
|---|---|---|
| Propranolol | Non-selective (β1+β2) | First β-blocker used; twice daily; largely replaced |
| Metoprolol | β1-selective | Preferred; reduces heart failure mortality; CYP2D6 metabolism |
| Atenolol | β1-selective | Renally excreted; once daily; less effective than metoprolol |
| Carvedilol | Non-selective + α1 | Also vasodilates; preferred in heart failure |
| Nebivolol | β1-selective + NO release | Vasodilatory; favorable metabolic profile |
Mechanism: Reduce CO (negative chronotropy/inotropy); reduce renin secretion (β1 in kidney); reduce central sympathetic outflow. Not first-line for uncomplicated hypertension but preferred with co-existing CAD, post-MI, HFrEF, or tachyarrhythmias.
Contraindications: Asthma (β2 blockade causes bronchoconstriction), severe bradycardia, heart block, decompensated heart failure.
Caution: Never stop abruptly - withdrawal syndrome (rebound tachycardia, angina, MI reported).
C. ACE Inhibitors (ACEi)
| Drug | Dose |
|---|---|
| Lisinopril | 10-40 mg/day |
| Ramipril | 2.5-10 mg/day |
| Enalapril | 5-40 mg/day |
| Captopril | 25-150 mg/day (TID) |
Mechanism: Block conversion of Ang I → Ang II → reduce vasoconstriction, reduce aldosterone release → reduce Na+/water retention. Also prevent bradykinin breakdown → increases NO and prostaglandin production (vasodilatory; also responsible for dry cough side effect).
Why favored: Particularly beneficial in diabetic nephropathy (reduce intraglomerular pressure), CKD with proteinuria, post-MI LV dysfunction, HFrEF.
Side effects: Dry cough (10-15%, due to bradykinin accumulation), angioedema (rare but serious), hyperkalemia, acute kidney injury in bilateral renal artery stenosis.
D. Angiotensin Receptor Blockers (ARBs)
| Drug | Dose |
|---|---|
| Losartan | 25-100 mg/day |
| Valsartan | 80-320 mg/day |
| Candesartan | 8-32 mg/day |
| Telmisartan | 20-80 mg/day |
Mechanism: Directly block AT1 receptors - prevent all Ang II effects (vasoconstriction, aldosterone release, sympathetic activation). Do NOT inhibit bradykinin breakdown, so no cough.
Key difference from ACEi: No cough; used when ACEi is not tolerated. Similar renoprotective and cardioprotective profile.
E. Calcium Channel Blockers (CCBs)
| Subclass | Examples | Primary Effect |
|---|---|---|
| Dihydropyridines (DHP) | Amlodipine, nifedipine, felodipine | Peripheral vasodilation (more selective); minimal cardiac depression |
| Non-DHP | Verapamil, diltiazem | Vasodilation + cardiac depression (reduce HR, conduction) |
Mechanism: Block L-type voltage-gated Ca2+ channels in vascular smooth muscle → vasodilation → reduced PVR. DHPs cause reflex sympathetic tachycardia (slight); non-DHPs slow heart rate and AV conduction.
Key uses: Amlodipine is a first-line agent for all patients. Preferred in African Americans, elderly, isolated systolic hypertension, angina. Non-DHPs are also used for rate control in AF.
Avoid: Non-DHPs with β-blockers (both depress heart → risk of heart block/bradycardia).
F. Other Drug Classes
| Drug | Class | Mechanism | Use |
|---|---|---|---|
| Clonidine | Central α2 agonist | Reduces central sympathetic outflow | Resistant hypertension; withdrawal |
| Hydralazine | Direct vasodilator | Increases cGMP → vascular smooth muscle relaxation | Heart failure (with nitrates); pregnancy |
| Minoxidil | Direct vasodilator | Opens K+ channels → hyperpolarization → vasodilation | Severe/resistant hypertension |
| Prazosin/doxazosin | α1 blocker | Blocks peripheral vasoconstriction | BPH + hypertension |
| Methyldopa | Central α2 agonist | First-line in pregnancy | Pregnancy hypertension |
| Nitroprusside | Direct vasodilator (NO) | Rapidly reduces both PVR and venous capacitance | Hypertensive emergency (IV) |
| Labetalol | α + β blocker | Combined effect | Hypertensive emergency; pregnancy |
| Nicardipine IV | CCB | Rapid vasodilation | Hypertensive emergency |
| Fenoldopam | DA1 agonist | Peripheral vasodilation + natriuresis | Hypertensive emergency |
6. Treatment Algorithm & Drug Selection Rationale
General Approach (Why we choose specific agents)
| Clinical Scenario | Preferred Drug(s) | Reason |
|---|---|---|
| Uncomplicated hypertension (first-line) | Thiazide, CCB, ACEi, or ARB | All proven to reduce CV events; choose based on comorbidities |
| Diabetes / CKD with proteinuria | ACEi or ARB | Reduce intraglomerular pressure; slow nephropathy progression |
| Post-MI / CAD / angina | β-blocker + ACEi/ARB | Reduce myocardial oxygen demand; prevent remodeling |
| Heart failure (HFrEF) | ACEi/ARB + β-blocker + aldosterone antagonist | RAAS blockade + sympatholysis + anti-fibrosis |
| Isolated systolic HTN in elderly | CCB (amlodipine) or thiazide | Well tolerated; efficacious in this group |
| African American patients | Thiazide or CCB (avoid β-blockers, ACEi as monotherapy) | β-blockers and ACEi less effective due to low-renin HTN |
| Asthma + HTN | Avoid β-blockers | β2 blockade → bronchoconstriction |
| Benign prostatic hyperplasia | α1 blocker (doxazosin) | Dual benefit: lower BP + relax prostate/bladder neck |
| Pregnancy | Methyldopa, labetalol, nifedipine | ACEi/ARBs are teratogenic (contraindicated) |
| Hyperaldosteronism / Resistant HTN | Spironolactone / eplerenone | Block excess mineralocorticoid effect |
Combination Therapy Rationale
When one drug is not sufficient (BP target ≥130/80 mmHg):
- Two-drug: ACEi/ARB + CCB or ACEi/ARB + thiazide
- Three-drug: ACEi/ARB + CCB + thiazide (most effective triple combo)
- Four-drug: Add spironolactone or β-blocker or clonidine for resistant hypertension
Fixed-dose triple combinations (e.g., telmisartan 20 mg + amlodipine 2.5 mg + chlorthalidone 12.5 mg) show high efficacy in moderate hypertension with minimal side effects and improve compliance.
BP Targets
- General population: <130/80 mmHg
- High CV risk / SPRINT trial supports: SBP target ~120 mmHg
- Elderly with isolated systolic HTN: treat if SBP >150 mmHg
- CKD with proteinuria: <130/80 mmHg
Causes of Treatment Failure (Resistant Hypertension)
- Medication non-compliance
- Excessive sodium intake
- Inadequate diuretic / volume overload
- Drug interactions: NSAIDs, TCAs, sympathomimetics, cocaine, caffeine, oral contraceptives
- Unidentified secondary cause (always re-assess)
7. Why Treat Hypertension? (The "Reason")
Untreated hypertension is the leading modifiable risk factor for:
- Stroke (both ischemic and hemorrhagic - 35-40% attributable to HTN)
- Myocardial infarction and coronary artery disease
- Heart failure (both HFrEF and HFpEF)
- Chronic kidney disease and ESRD (second leading cause after diabetes)
- Aortic aneurysm and dissection
- Peripheral artery disease (2.5-4x increased risk)
- Retinopathy and vision loss
- Vascular dementia
Treatment reduces:
- Stroke risk by ~35-40%
- Coronary events by ~20-25%
- Heart failure by ~50%
- Cardiovascular mortality significantly
Sources: Fuster & Hurst's The Heart 15th ed. | Katzung's Basic & Clinical Pharmacology 16th ed. | Comprehensive Clinical Nephrology 7th ed. | Brenner & Rector's The Kidney | Goldman-Cecil Medicine
Recent evidence note: A 2025 systematic review [PMID 40885884] highlights aldosterone synthase inhibitors as emerging agents for resistant hypertension, adding to the existing drug classes above.
Psoriasis physiology, pathology pharmacology causes
Reading File
Finding Sources
Searching PubMed
"psoriasis" AND "pathophysiology"
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive data from all the best sources. Let me compile the complete answer.
Psoriasis: Complete Overview
1. Definition & Epidemiology
Psoriasis is a common, chronic, immune-mediated inflammatory skin disease affecting 1-2% of the world's population. It is characterized by discrete, well-demarcated erythematous plaques covered by silvery-white micaceous scale. It is now recognized as a systemic disease - not merely a skin condition - with significant cardiovascular, metabolic, and articular comorbidities that increase morbidity and reduce life expectancy.
2. Causes & Risk Factors
Psoriasis results from an interplay of genetic susceptibility and environmental triggers.
A. Genetic Factors
- HLA association: HLA-Cw6 (also written HLA-C*06:02) is the strongest genetic risk factor - present in ~60-65% of early-onset psoriasis patients
- GWAS findings: Susceptibility loci involve genes for:
- Adaptive immunity (T-cell activation pathways)
- TNF signaling (TNFA gene polymorphisms)
- Skin barrier function
- IL-23/IL-17 axis regulation
- Genetic susceptibility loci for psoriasis are largely non-overlapping with those for atopic dermatitis, confirming distinct pathogenic mechanisms
- First-degree relatives have ~10-fold increased risk
B. Environmental Triggers
| Trigger | Mechanism |
|---|---|
| Streptococcal pharyngitis | Most strongly linked to guttate psoriasis; streptococcal superantigens activate T cells |
| Stress | HPA axis activation, neuropeptide release, immune dysregulation |
| Physical trauma | Koebner (isomorphic) phenomenon - plaques form at sites of skin injury |
| Drugs | Lithium, beta-blockers, antimalarial drugs (chloroquine), NSAIDs, systemic steroid withdrawal |
| Infections | HIV (can trigger severe/atypical psoriasis), fungal infections |
| Obesity | Metabolic inflammation drives cytokine production; visceral fat is proinflammatory |
| Alcohol & smoking | Both associated with increased severity and treatment resistance |
| Pregnancy | Can exacerbate or improve psoriasis (unpredictable); pustular psoriasis risk |
| Sunburn | Paradoxically can trigger new lesions via Koebner phenomenon |
3. Physiology / Pathophysiology
Psoriasis involves a self-perpetuating cycle of immune activation and keratinocyte hyperproliferation, centered on the IL-23/IL-17 axis.
Step-by-step Immunological Cascade:
Step 1 - Innate triggering (initiating event)
An unknown trigger (possibly microbial antigens, self-peptides, or stress signals) activates plasmacytoid dendritic cells (pDCs) and myeloid dendritic cells (mDCs) in the dermis. These DCs produce:
- TNF-α - primary proinflammatory cytokine
- IL-23 - the critical "master regulator" - a heterodimer (p19 + p40 subunits)
- IL-12 - promotes Th1 differentiation (shares p40 subunit with IL-23)
Step 2 - Adaptive immune activation (T-cell polarization)
- IL-23 drives differentiation and survival of Th17 cells
- Th17 cells produce IL-17A, IL-17F, IL-22, IL-21
- IL-12 drives Th1 cells → produce IFN-γ
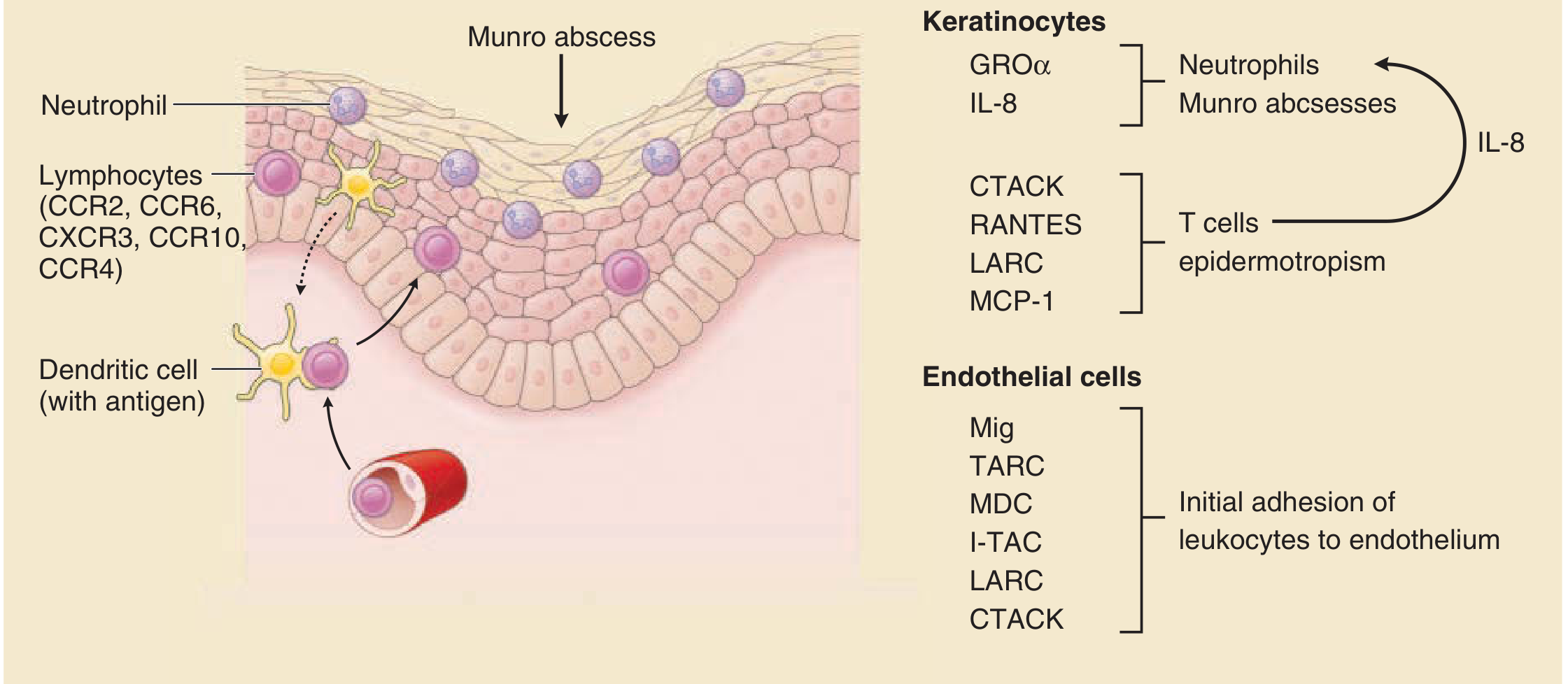
- Both Th1 and Th17 memory T cells home to skin via chemokine gradients (CTACK, RANTES, LARC, MCP-1) and chemokine receptors (CCR2, CCR6, CXCR3, CCR10, CCR4)
Step 3 - Keratinocyte hyperproliferation
- IL-17 acts on keratinocytes to:
- Massively upregulate antimicrobial peptides (defensins, LL-37)
- Increase chemokine production (IL-8/CXCL8, GRO-α) → recruit neutrophils
- Drive keratinocyte proliferation
- IL-22 acts on keratinocytes to cause acanthosis (epidermal thickening)
- TNF-α augments all IL-17 effects on keratinocytes - creating a feed-forward inflammatory amplification loop
- Normal keratinocyte transit time from basal layer to surface = ~28 days; in psoriasis this is reduced to 3-5 days - cells don't mature properly
Step 4 - Vascular remodeling ("squirting papillae")
Capillary loops in dermal papillae:
- Elongate, dilate, and remodel their basement membrane to resemble postcapillary venules
- Express adhesion molecules (E-selectin, ICAM-1) supporting leukocyte extravasation
- Leukocytes (lymphocytes + neutrophils) can now exit directly within papillary tips into the epidermis - the "squirting papillae" sign unique to psoriasis
Step 5 - Neutrophil recruitment
- IL-8 and GRO-α produced by keratinocytes recruit neutrophils into the epidermis
- Neutrophils aggregate in the stratum corneum → Munro microabscesses
- Neutrophils aggregate in the spinous layer → spongiform pustules of Kogoj (especially in pustular psoriasis)
The IL-17/TNF feed-forward loop (key concept):
TNF-α + IL-17 → keratinocytes produce more IL-8 → more neutrophils → more inflammation → more cytokine release → sustained chronic inflammation
4. Pathology (Histopathology)
Gross Appearance
- Well-demarcated pink/salmon-red plaques with loosely adherent silver-white (micaceous) scale
- In darker skin types: hyperpigmented plaques with gray scale
- Auspitz sign: punctate bleeding points when scale is removed (exposed dilated capillary tips)
Microscopic Features (H&E)
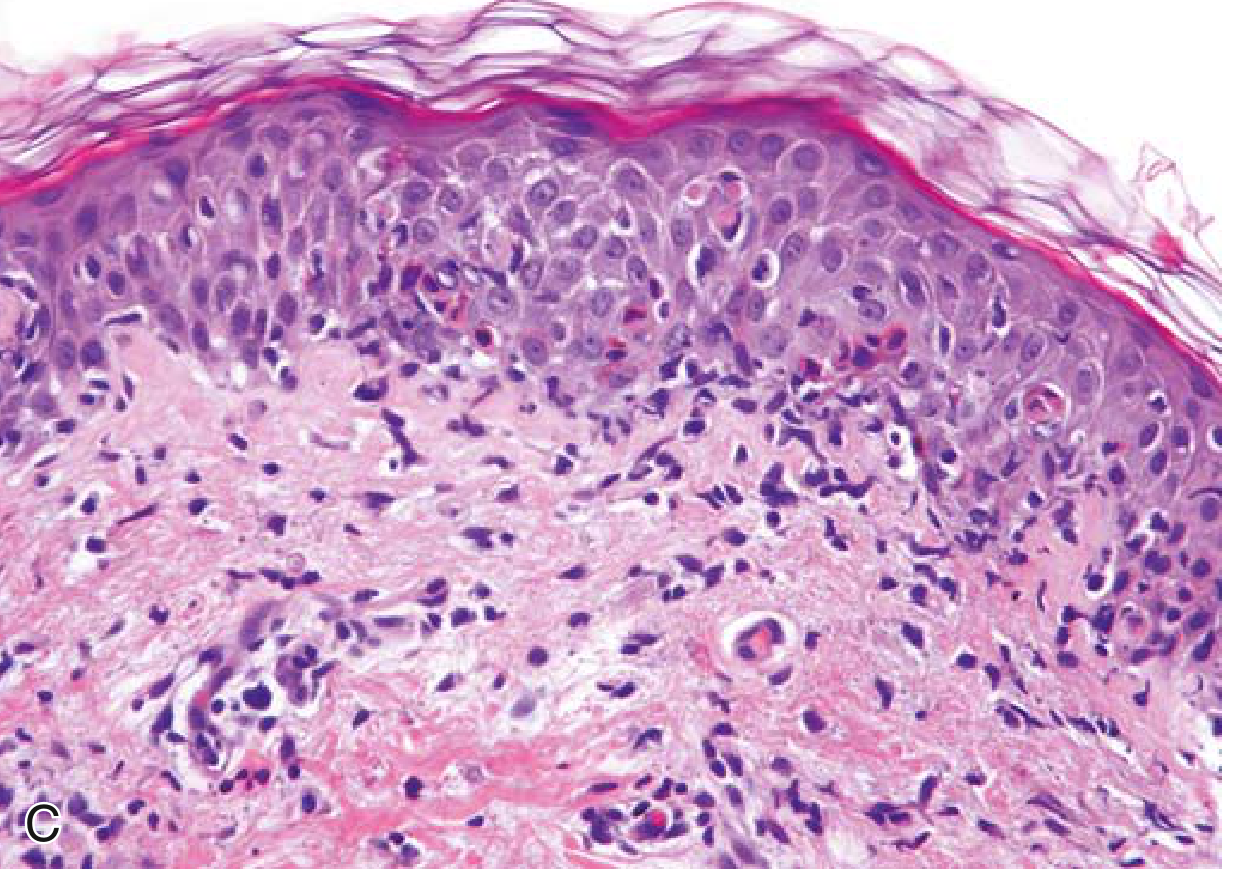
| Feature | Description |
|---|---|
| Acanthosis | Marked epidermal thickening with regular elongation of rete ridges ("test tubes in a rack") |
| Parakeratosis | Retention of nuclei in stratum corneum (cells don't mature = keratinocytes shed nuclei too late) |
| Loss of stratum granulosum | Due to rapid cell turnover and abnormal differentiation |
| Hypogranulosis | Reduced/absent granular layer |
| Dilated and tortuous capillaries | Within dermal papillae - elongated, reaching up to the epidermis |
| Thinning of suprapapillary epidermis | Epidermis over tips of papillae is paradoxically thin despite overall thickening |
| Munro microabscesses | Collections of neutrophils in the parakeratotic stratum corneum |
| Spongiform pustule of Kogoj | Neutrophil aggregates within the spinous layer (especially pustular psoriasis) |
| Mixed dermal infiltrate | Predominantly CD4+ Th1/Th17 T cells, macrophages, dendritic cells; neutrophils |
Clinical Variants and Their Pathology
| Variant | Key Features |
|---|---|
| Plaque psoriasis (psoriasis vulgaris) | Most common (80-90%); chronic stable plaques; elbows, knees, scalp, lumbosacral |
| Guttate psoriasis | Small "raindrop" papules; acute onset; often post-streptococcal; younger patients |
| Pustular psoriasis | Sterile pustules on erythematous skin; generalized form has fever; triggered by steroid withdrawal |
| Erythrodermic psoriasis | >90% BSA involvement; life-threatening; thermoregulation failure, high-output cardiac failure risk |
| Inverse psoriasis | Intertriginous areas (axilla, groin, submammary); moist, minimal scale |
| Nail psoriasis | Pitting, onycholysis, subungual hyperkeratosis, "oil drop" sign; seen in 30% |
| Psoriatic arthritis (PsA) | Up to 30% of patients; seronegative; five subtypes (see below) |
Psoriatic Arthritis Subtypes
- Symmetric PsA (~50%): resembles RA
- Asymmetric PsA (~35%): "sausage digits" (dactylitis)
- Distal PsA (~5%): classic, DIP joints with nail disease
- Spondylitis (~5%): axial involvement
- Arthritis mutilans (<5%): severe destructive, "pencil-in-cup" deformity
5. Pharmacology & Treatment
Treatment is stratified by severity using the PASI score (Psoriasis Area and Severity Index) and BSA (Body Surface Area).
STEP 1: Topical Therapy (Mild-Moderate Disease)
| Drug | Class | Mechanism | Notes |
|---|---|---|---|
| Corticosteroids (e.g., betamethasone, clobetasol) | Anti-inflammatory | Inhibit phospholipase A2, reduce cytokine production, vasoconstriction | First-line for most patients; high-potency for body; low-potency for face/flexures |
| Calcipotriol (calcipotriene) | Vitamin D3 analogue | Binds VDR → inhibits keratinocyte proliferation, promotes differentiation, immunomodulatory | Effective; combined with betamethasone in "Dovobet" (synergistic) |
| Tazarotene | Topical retinoid | Binds RAR → normalizes keratinocyte differentiation, anti-proliferative | Effective; can be irritating; teratogenic (pregnancy Category X) |
| Coal tar | Keratolytic/antipruritic | Reduces epidermal proliferation; mechanism not fully understood | Old but effective; smelly; photosensitizing |
| Salicylic acid | Keratolytic | Reduces scale by disrupting corneocyte attachments | Used as adjunct to enhance penetration of other drugs |
| Anthralin (dithranol) | Antiproliferative | Inhibits mitochondrial function in keratinocytes | Effective; stains skin and clothing |
| Tacrolimus / pimecrolimus | Calcineurin inhibitor (topical) | Inhibit T-cell activation; no skin atrophy | Useful for face, flexures; not first-line |
STEP 2: Phototherapy (Moderate-Severe Disease)
| Type | Mechanism | Notes |
|---|---|---|
| Narrowband UVB (NB-UVB) | Immunosuppressive; induces T-cell apoptosis, reduces cytokines | Preferred phototherapy; 3x/week; safer than PUVA |
| PUVA (psoralen + UVA) | Psoralens intercalate DNA → UVA induces cross-links → anti-proliferative | Effective but increased melanoma/non-melanoma skin cancer risk; contraindicated with cyclosporine |
| Broadband UVB | Less selective than NB-UVB | Less commonly used now |
STEP 3: Systemic Non-biologic Agents
| Drug | Class | Mechanism | Dose | Key Side Effects |
|---|---|---|---|---|
| Methotrexate | Antimetabolite (DMARD) | Inhibits dihydrofolate reductase → blocks DNA synthesis in rapidly dividing cells (keratinocytes + lymphocytes); anti-inflammatory via adenosine release | 7.5-25 mg/week (oral or SC) | Hepatotoxicity, bone marrow suppression, teratogenicity, pneumonitis; supplement folic acid |
| Cyclosporine | Calcineurin inhibitor | Binds cyclophilin → inhibits calcineurin → blocks NFAT → reduces IL-2, T-cell activation | 3-5 mg/kg/day | Nephrotoxicity, hypertension, infections, malignancy risk; use short-term |
| Acitretin | Oral retinoid (systemic) | Binds nuclear retinoic acid receptors (RARs) → normalizes keratinocyte differentiation | 25-50 mg/day | Teratogenic (3-year contraception after stopping); dyslipidemia; mucocutaneous dryness; preferred when immunosuppression must be avoided |
| Apremilast | PDE-4 inhibitor (small molecule) | Inhibits phosphodiesterase 4 → raises intracellular cAMP → reduces TNF-α, IL-17, IL-23 production | 30 mg twice daily (after titration) | Diarrhea, nausea, depression; dose reduce in renal failure; no immunosuppression risk |
| Deucravacitinib | TYK2 inhibitor | Inhibits tyrosine kinase 2 → blocks IL-23, IL-12, type-I interferon signaling | 6 mg daily | Oral; approved for moderate-severe plaque psoriasis; favorable safety vs JAK inhibitors |
CAUTION: Oral glucocorticoids are contraindicated in psoriasis - withdrawal can precipitate life-threatening generalized pustular psoriasis (von Zumbusch variant).
STEP 4: Biologic Agents (Moderate-Severe Disease)
The development of biologics transformed psoriasis management. Each class targets a specific cytokine proven essential in psoriasis pathogenesis:
a) Anti-TNF-α Agents
Rationale: TNF-α is a primary driver of inflammation; elevated in psoriatic skin and synergizes with IL-17.
| Drug | Route | Notes |
|---|---|---|
| Adalimumab (Humira) | SC | Human anti-TNF-α mAb; also approved for PsA |
| Etanercept (Enbrel) | SC | TNF-receptor fusion protein; milder effect than mAbs |
| Infliximab (Remicade) | IV | Chimeric mAb; rapid onset; effective in severe disease |
| Certolizumab (Cimzia) | SC | PEGylated Fab fragment; preferred in pregnancy (minimal placental transfer) |
| Golimumab (Simponi) | SC | Approved for PsA only |
Warnings: Serious infections (TB reactivation - screen before starting), hepatotoxicity, worsening CHF, hematologic events, risk of demyelinating disease, potential increased malignancy.
b) Anti-IL-12/IL-23 (p40 Subunit)
| Drug | Route | Notes |
|---|---|---|
| Ustekinumab (Stelara) | SC | Human mAb targeting p40 (shared subunit of IL-12 and IL-23); every 12 weeks after loading; effective in psoriasis and PsA |
c) Anti-IL-23 (p19 Subunit) - Most Selective
Rationale: IL-23 is the upstream "master regulator" of Th17 differentiation; more selective than p40 inhibition.
| Drug | Route | Notes |
|---|---|---|
| Guselkumab (Tremfya) | SC | IL-23 p19 specific; high efficacy |
| Risankizumab (Skyrizi) | SC | IL-23 p19 specific; every 12 weeks |
| Tildrakizumab (Ilumya) | SC | IL-23 p19 specific |
d) Anti-IL-17 Agents - Fastest/Highest Efficacy
Rationale: IL-17A is the direct effector cytokine causing keratinocyte hyperproliferation; blocking it gives dramatic responses (often complete clearance).
| Drug | Target | Route | Notes |
|---|---|---|---|
| Secukinumab (Cosentyx) | IL-17A | SC | First anti-IL-17A; remarkable efficacy; rapid onset |
| Ixekizumab (Taltz) | IL-17A | SC | Similar to secukinumab; very high PASI 90/100 response rates |
| Brodalumab (Siliq) | IL-17 receptor A | SC | Blocks all IL-17 isoforms; black box warning for suicidal ideation |
Side effect note for IL-17 blockers: Increased risk of Candida infections (IL-17 is important for mucosal antifungal immunity); avoid in inflammatory bowel disease (can worsen it).
Drug Selection Summary
| Scenario | Preferred Treatment |
|---|---|
| Mild (BSA <3%, PASI <5) | Topical corticosteroid ± calcipotriol |
| Scalp psoriasis | High-potency topical steroid (foam/solution) + calcipotriol |
| Nail psoriasis | Topical steroids, calcipotriol; biologics if severe |
| Moderate (BSA 3-10%) | NB-UVB phototherapy or topical + one systemic |
| Severe (BSA >10%, PASI >10) | Methotrexate, cyclosporine, or biologic |
| Psoriatic arthritis | Methotrexate + anti-TNF or anti-IL-17 (anti-IL-17 preferred for skin + joints) |
| Pregnancy | Topical steroids (low-to-mid potency), NB-UVB; certolizumab if biologic needed |
| Rapid response needed | Cyclosporine (fastest systemic) or infliximab IV |
| Avoid immunosuppression | Acitretin or NB-UVB |
| When anti-TNF inadequate | Switch to anti-IL-17 or anti-IL-23 (higher efficacy) |
| Avoid injections (mild-mod) | Apremilast or deucravacitinib (oral small molecules) |
6. Comorbidities (The Systemic Disease Concept)
Psoriasis is a systemic inflammatory disease - chronic immune activation affects multiple organ systems:
| Comorbidity | Risk/Notes |
|---|---|
| Psoriatic arthritis | ~30% of patients; often undertreated |
| Cardiovascular disease | Accelerated atherosclerosis; increased MI, stroke risk |
| Metabolic syndrome | Obesity, insulin resistance, dyslipidemia, hypertension |
| Depression/anxiety | Due to disease burden + shared neuroinflammatory mechanisms |
| Non-alcoholic fatty liver disease | Especially with methotrexate use (hepatotoxic) |
| Inflammatory bowel disease | ~7% prevalence; anti-IL-17 drugs are contraindicated |
| Malignancy | Lymphoma risk (especially with PUVA); squamous cell carcinoma |
Sources: Fitzpatrick's Dermatology (Volumes 1 & 2) | Robbins & Kumar Basic Pathology | Harrison's Principles of Internal Medicine 22nd ed. | Katzung's Basic and Clinical Pharmacology 16th ed.
Recent evidence note: A 2025 systematic review [PMID 39960751] highlights emerging pathophysiological links between psoriasis and type 2 diabetes, suggesting shared inflammatory mediators (TNF-α, IL-17, IL-6) may explain the concurrent metabolic dysregulation seen in psoriatic patients - supporting early metabolic screening in all psoriasis patients.
This is a shared conversation. Sign in to Orris to start your own chat.