Pathogenesis of sickle cell disease. Pathology 5-mark question
"sickle cell disease"[MeSH Terms] AND pathogenesis
Pathogenesis of Sickle Cell Disease
Genetic Basis
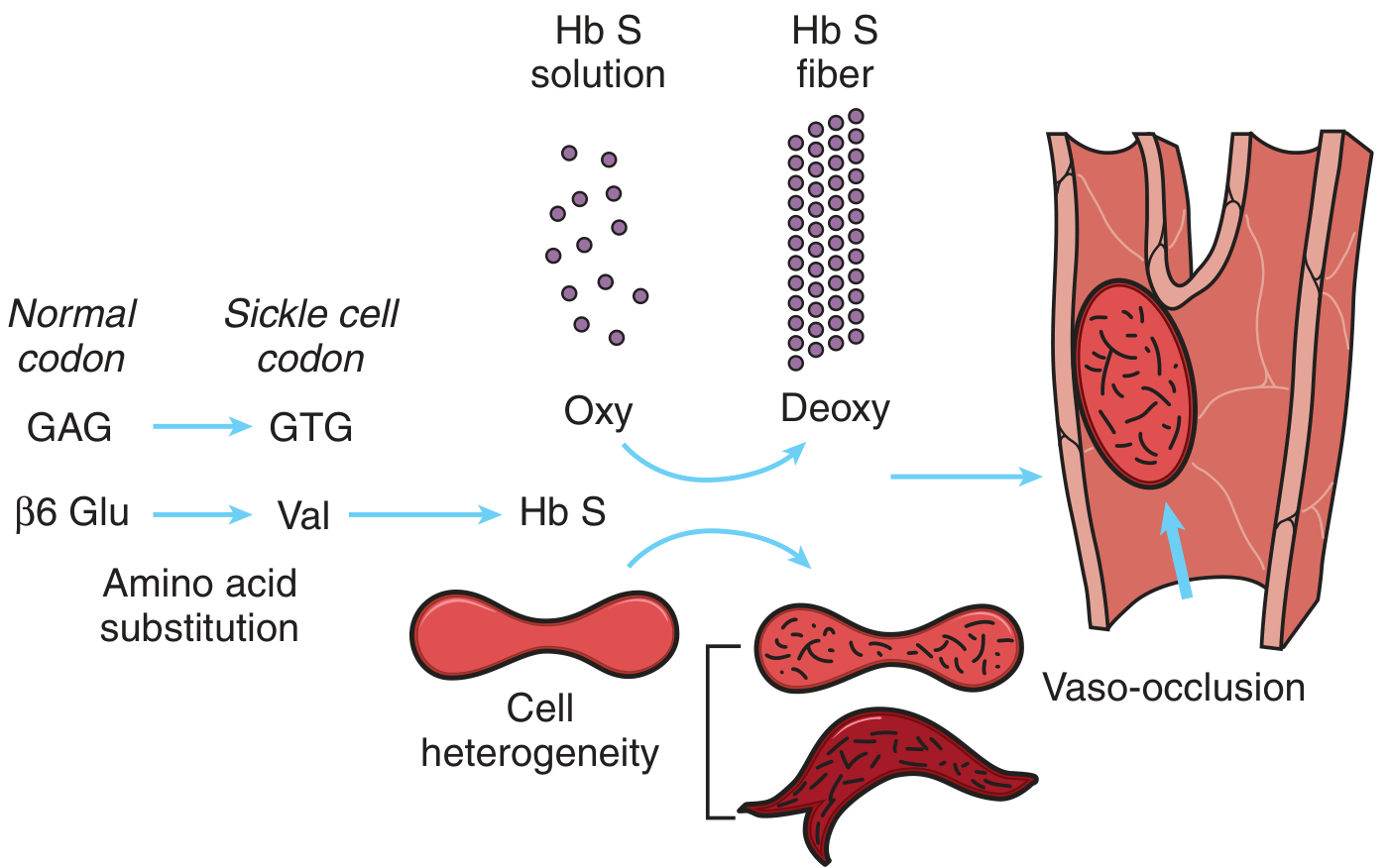
Step-by-Step Pathogenetic Cascade
1. HbS Polymerization Under Hypoxia
- In the oxygenated state, HbS functions almost normally (solubility is close to HbA).
- Under deoxygenated conditions, HbS becomes only one-fifth as soluble as HbA.
- The hydrophobic valine residue creates a complementary "sticky patch" that allows deoxygenated HbS molecules to undergo intermolecular contacts and aggregate into rod-shaped polymers/fibers.
- These polymers distort the red cell membrane, producing the elongated, crescentic sickle shape.
2. Reversible vs. Irreversible Sickling
- Early sickling episodes are reversible upon reoxygenation.
- However, repeated cycles of sickling cause calcium influx into the red cell, leading to loss of potassium and water (cellular dehydration) and progressive membrane skeleton damage.
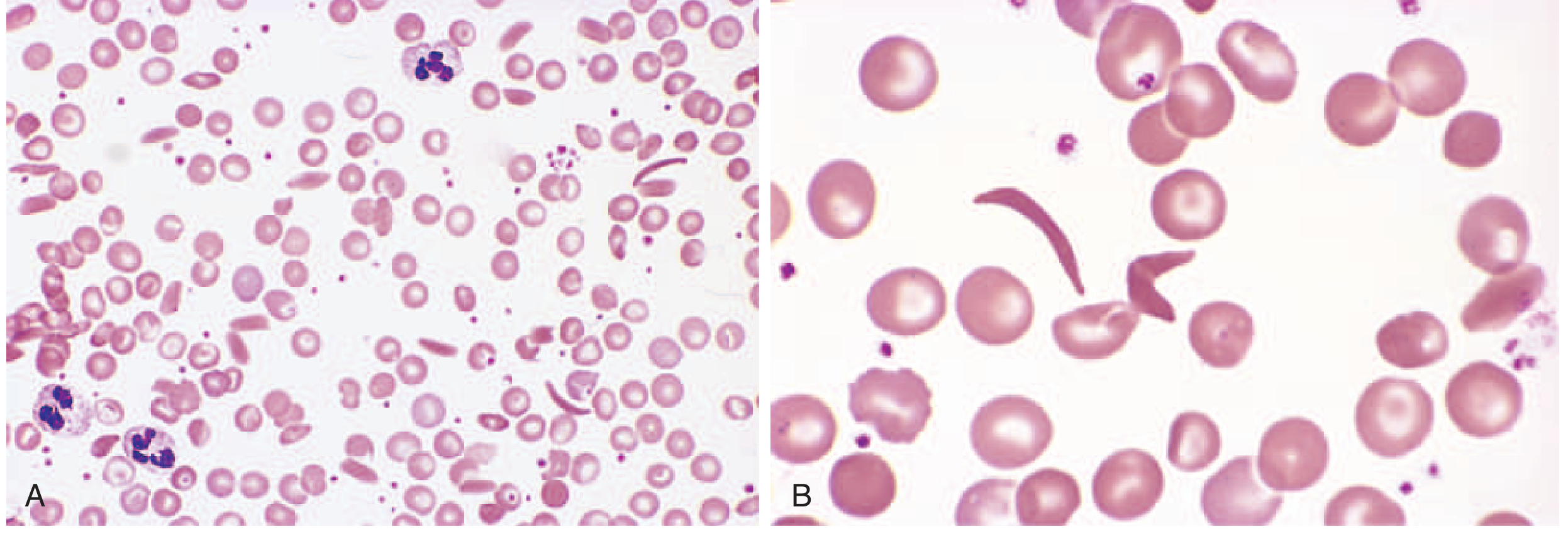
- Over time, this cumulative injury creates irreversibly sickled cells (ISCs) - these cannot regain normal shape even upon reoxygenation, and are prone to intravascular hemolysis.
3. Factors Governing Clinical Severity of Sickling
| Factor | Effect |
|---|---|
| Intracellular concentration of non-HbS hemoglobin | HbA and HbF both inhibit HbS polymerization. In heterozygotes (~40% HbS), sickling is rare. High HbF levels (as in neonates) protect until ~5-6 months of age. |
| Intracellular HbS concentration | Dehydration increases Hb concentration, promoting polymerization. Co-existing α-thalassemia reduces Hb concentration and is protective. |
| Microvascular transit time | Normal capillary transit is too brief for significant polymerization. Sluggish flow (spleen, bone marrow) or factors like infection/inflammation prolong transit, enabling sickling. |
4. Two Major Pathological Consequences
A. Hemolytic Anemia
- Membrane damage from repeated sickling reduces red cell lifespan to ~20 days (normal: ~120 days) - one-sixth of normal.
- Leads to moderate-to-severe normochromic, normocytic hemolytic anemia.
- Severity correlates with the fraction of irreversibly sickled cells in blood.
- Hemolysis releases free hemoglobin, which scavenges nitric oxide (NO), impairing vasodilation and worsening ischemia.
B. Vaso-occlusion and Ischemic Injury
- Sickle cells physically obstruct the microvasculature.
- Repeated sickling makes the red cell membrane abnormally "sticky" (increased adhesion to endothelium).
- Vaso-occlusion is not directly proportional to ISC numbers; it is triggered by precipitating factors: infection, inflammation, dehydration, acidosis, hypoxia, and cold.
- Inflammation slows blood flow further by increasing leukocyte-endothelial adhesion and leaky vessels - creating a vicious cycle.
Pathological Changes (Morphology)
| Organ/System | Change |
|---|---|
| Spleen | Initially congestive splenomegaly (up to 500 g in children) → repeated infarcts → autosplenectomy (fibrotic nubbin) by adulthood |
| Peripheral blood smear | Elongated, boat-shaped sickled cells, anisocytosis, poikilocytosis, target cells |
| Bone marrow | Compensatory erythroid hyperplasia → bone resorption + new bone formation → "crew-cut" pattern on skull X-ray, prominent cheekbones |
| Multiple organs | Vascular thrombosis and infarction in bones, kidneys, brain, retina, lungs, skin |
| Liver/Heart | Fatty changes from hypoxia-induced ischemia |
| Gallbladder | Pigment gallstones (from chronic hemolysis and bilirubin excess) |
| Kidney | Papillary necrosis, hematuria, hyposthenuria |
| Penis | Priapism → penile fibrosis, erectile dysfunction |
Role of Modifier Genes (HbF)
- HbF (α₂γ₂) inhibits HbS polymerization by failing to form intermolecular contacts with deoxygenated HbS.
- Higher HbF = less clinical severity (reduced pain crises, longer survival).
- BCL11A gene product is a transcription factor that silences γ-globin expression postnatally. Loss-of-function of BCL11A → hereditary persistence of fetal hemoglobin (HPFH), which ameliorates sickle cell disease.
- Hydroxyurea therapy works by increasing HbF levels (via a poorly understood mechanism), reducing sickling frequency.
Summary (5 Key Points for Exam)
- Molecular defect: GAG→GTG mutation → Glu→Val at β-globin codon 6 → HbS
- Polymerization: Deoxygenated HbS forms rigid polymers → red cell deforms into sickle shape
- Reversible → Irreversible: Repeated sickling causes Ca²⁺ influx, membrane damage, irreversibly sickled cells
- Hemolysis: RBC lifespan reduced to ~20 days; free Hb scavenges NO → worsens ischemia
- Vaso-occlusion: Sticky sickled cells + sluggish flow → microvascular obstruction → ischemia, infarction, pain crises (bones, spleen, brain, kidney)
Pathogenesis of sickle cell disease. Pathology 5-mark question. Give in flow chats
FLOW CHART 1: Molecular Basis (The Core Defect)
Point mutation in β-globin gene (chromosome 11)
↓
GAG → GTG (codon 6)
↓
Glutamic acid → Valine substitution
(hydrophilic) (hydrophobic)
↓
Abnormal hemoglobin = HbS (α₂β^S₂)
↓
In homozygotes (HbSS): HbA completely replaced by HbS
↓
SICKLE CELL DISEASE
FLOW CHART 2: Sickling Mechanism
HbS
↓
┌───────────────────────┐
↓ ↓
OXYGENATED state DEOXYGENATED state
(normal solubility) (1/5th solubility of HbA)
↓ ↓
No sickling Hydrophobic valine creates
"sticky patches" on HbS
↓
HbS molecules undergo conformational change
↓
Intermolecular contacts → Rod-shaped
HbS polymers/fibers form
↓
Distortion of red cell membrane
↓
Elongated, crescentic SICKLE SHAPE
FLOW CHART 3: Reversible → Irreversible Sickling
First few sickling episodes
↓
REVERSIBLE sickling
(re-oxygenation restores shape)
↓
Repeated cycles of sickling
↓
Ca²⁺ influx into red cell
↓
Loss of K⁺ and water → Cell dehydration
↓
Membrane skeleton damage (cumulative)
↓
IRREVERSIBLY SICKLED CELLS (ISCs)
(cannot regain normal shape even on re-oxygenation)
↓
Prone to intravascular HEMOLYSIS
FLOW CHART 4: Two Major Pathological Arms
SICKLED RED CELLS
↓
┌──────────────────────────────┐
↓ ↓
ARM 1: HEMOLYSIS ARM 2: VASO-OCCLUSION
↓ ↓
RBC lifespan ↓ to ~20 days Sticky sickled cells adhere
(normal = 120 days) to vascular endothelium
↓ ↓
Hemolytic anemia Microvascular obstruction
(moderate to severe) ↓
↓ Ischemia → Infarction
Free Hb released ↓
↓ PAIN CRISES + organ damage
Scavenges Nitric Oxide (NO)
↓
Impaired vasodilation
→ Worsens ischemia
FLOW CHART 5: Vaso-occlusion in Detail
Precipitating factors:
Infection / Inflammation / Dehydration / Acidosis / Hypoxia / Cold
↓
Sluggish blood flow in microvasculature
(especially spleen, bone marrow)
↓
Prolonged transit time of sickle RBCs
↓
More time for HbS polymerization to occur
↓
Increased RBC adhesion to endothelium
(membrane damage → abnormally "sticky" cells)
↓
Leukocyte adhesion + plasma exudation
(inflammation worsens flow)
↓
┌─────────────────────────────────────────┐
↓ ↓ ↓ ↓
Bones Spleen Brain Kidney
(infarcts) (autosplenectomy) (stroke) (papillary necrosis)
↓ ↓ ↓ ↓
Lung Retina Liver Penis
(acute chest) (blindness) (ischemia) (priapism)
FLOW CHART 6: Splenic Changes (Classic Sequence)
Childhood
↓
Sickled RBCs trapped in splenic red pulp
↓
Congestive SPLENOMEGALY (up to 500 g)
↓
Repeated hypoxic damage + infarcts
↓
Progressive fibrosis and shrinkage
↓
AUTOSPLENECTOMY (by adulthood)
(small, fibrotic, non-functional nubbin)
↓
Functional asplenia
↓
Susceptibility to encapsulated organisms
(Streptococcus pneumoniae, H. influenzae, Salmonella)
FLOW CHART 7: Role of HbF (Modifier Gene)
BCL11A gene → silences γ-globin → ↓ HbF postnatally
↓
Low HbF → HbS polymerization proceeds freely
↓
Severe disease
─────────────────────────────────
High HbF (e.g., HPFH, hydroxyurea therapy)
↓
HbF interacts weakly with deoxygenated HbS
↓
Inhibits HbS polymer formation
↓
↓ Sickling → ↓ Pain crises → Milder disease
MASTER SUMMARY FLOW CHART
β-globin gene mutation (Glu6Val)
↓
HbS
↓
Deoxygenation in tissues
↓
HbS polymerization → Fiber formation
↓
Red cell sickling
↙ ↘
HEMOLYSIS VASO-OCCLUSION
↓ ↓
Anemia Ischemia & Infarction
↓ ↓
Jaundice Pain crises, Organ damage
Pigment stones Stroke, Autosplenectomy
Acute chest syndrome
Create a visual infographic poster of sickle cell pathogenesis flowchart
A detailed medical education infographic poster titled "Pathogenesis of Sickle Cell Disease" with a dark navy blue background and bright colored flowchart elements. The poster is organized top to bottom with connecting arrows showing the disease cascade: TOP SECTION - "Genetic Defect" box in red: β-globin gene mutation on chromosome 11, GAG→GTG (codon 6), Glutamic Acid → Valine substitution, producing HbS (abnormal hemoglobin) SECOND SECTION - "HbS Polymerization" box in orange: shows two pathways side by side - left path labeled "Oxygenated" showing normal round red blood cells, right path labeled "Deoxygenated" showing HbS molecules forming rod-shaped polymers and fibers, with a large arrow pointing right labeled "Low O2 / Hypoxia" THIRD SECTION - "Sickling" box in yellow: illustration of red blood cells distorting into elongated crescent sickle shapes, with text "Reversible at first → Repeated cycles → Ca2+ influx → Membrane damage → IRREVERSIBLY SICKLED CELLS" FOURTH SECTION splits into TWO parallel arms with a large downward arrow: LEFT ARM - "Hemolysis" box in purple: RBC lifespan 20 days (normal 120), Hemolytic anemia, Free Hb released → Scavenges Nitric Oxide → Impaired vasodilation → Worsened ischemia RIGHT ARM - "Vaso-Occlusion" box in teal/cyan: Sticky sickled cells adhere to endothelium, Microvascular obstruction, Precipitated by: Infection, Dehydration, Acidosis, Cold, Hypoxia BOTTOM SECTION - "Organ Damage" box in coral red showing 6 organ icons with labels: Spleen (Autosplenectomy), Brain (Stroke), Bone (Infarcts/Crewcut skull), Lung (Acute chest syndrome), Kidney (Papillary necrosis), Penis (Priapism) BOTTOM BANNER in gold: "HbF is PROTECTIVE - inhibits HbS polymerization | Hydroxyurea increases HbF" Clean medical illustration style with white text, connecting arrows in bright white and yellow, subtle red blood cell illustrations as background watermarks, professional clinical poster layout
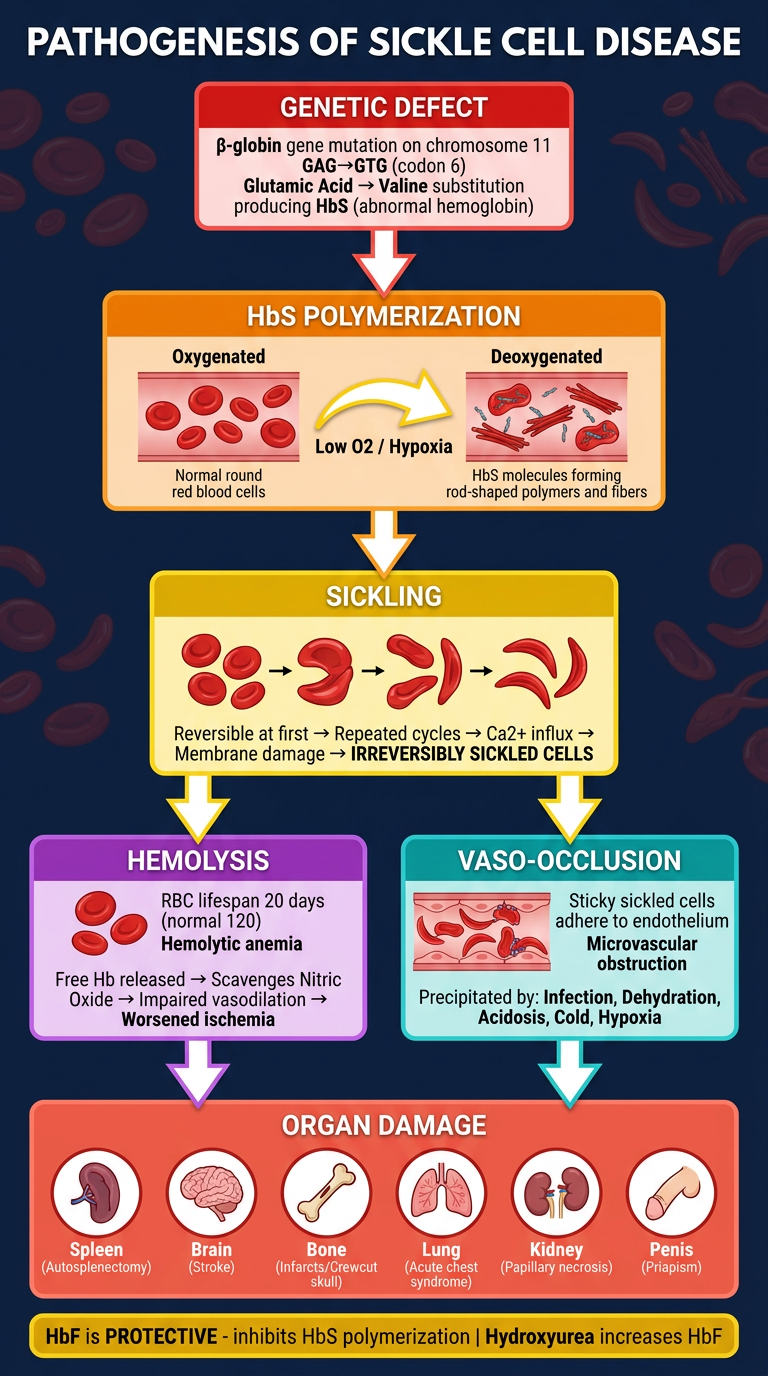
- Top - the genetic root cause (GAG→GTG mutation → Glu→Val → HbS)
- Middle - HbS polymerization under deoxygenation → sickling (reversible → irreversible)
- Split arms - the two major consequences: Hemolysis (left) and Vaso-occlusion (right)
- Bottom - end-organ damage across 6 systems (spleen, brain, bone, lung, kidney, penis)
- Footer banner - the protective role of HbF and hydroxyurea therapy