Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
I now have all the material needed for a thorough, well-illustrated response. Let me compose the answer.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
ARDS is a clinical syndrome of severe, rapid-onset respiratory failure caused by diffuse alveolar damage (DAD) from a wide range of direct and indirect pulmonary insults. The mechanism unfolds through three overlapping phases: exudative, proliferative, and fibrotic.
Triggers and Initial Insult
The most common causes are pneumonia and sepsis (~40-60%), followed by aspiration, major trauma, and multiple transfusions. Lung injury can be direct (e.g., toxic inhalation, pneumonia, aspiration) or indirect (e.g., sepsis, pancreatitis, severe burns), with the end result being the same pathologic cascade.
- Harrison's Principles of Internal Medicine, 22nd ed., Ch. 312
Phase 1: Exudative Phase (Days 0-7)
This is the core injurious mechanism:
1. Barrier disruption
The alveolar-capillary barrier is formed by two layers: capillary endothelial cells and alveolar epithelial cells (type I pneumocytes cover ~95% of the alveolar surface). In ARDS, both layers are injured, destroying the normally tight barrier to fluid and macromolecules.
2. Inflammatory cascade activation
Toll-like receptors on alveolar type I (ATI) epithelial cells and resident alveolar macrophages detect the initial insult (bacteria, viruses, DAMPs from trauma) and secrete proinflammatory cytokines - principally IL-1, IL-6, IL-8, and TNF-α - along with lipid mediators like leukotriene B4. These recruit circulating leukocytes, predominantly neutrophils, into the pulmonary interstitium and alveoli.
3. Neutrophil-mediated injury (central mechanism)
Neutrophils are the key effectors of alveolar injury. As they migrate across the epithelium, they release:
- Proteases (elastase, matrix metalloproteinases) that digest structural proteins
- Reactive oxygen species (ROS) causing oxidative damage
- Neutrophil extracellular traps (NETs) - webs of chromatin and antimicrobial proteins that also exacerbate endothelial and epithelial injury
4. Monocyte and platelet involvement
Monocytes recruited into the lung can trigger epithelial cell apoptosis via IFN-β-dependent release of TRAIL (TNF-related apoptosis-inducing ligand), which activates death receptors. Activated platelets form aggregates with PMN leukocytes (relevant to NET formation). Red blood cells leak into the alveolar space; cell-free hemoglobin released from RBCs exacerbates injury via oxidant-dependent mechanisms.
5. Protein-rich alveolar flooding
The disrupted barrier allows protein-rich edema fluid to accumulate in the interstitium and alveolar spaces. Condensed plasma proteins combine with cellular debris and dysfunctional surfactant to form the characteristic hyaline membranes (eosinophilic deposits lining the alveolar walls on histology).
6. Surfactant failure
Phospholipase A2 (released in pancreatitis-associated ARDS and other states) enzymatically degrades surfactant. Loss of surfactant increases alveolar surface tension, promoting alveolar collapse and further worsening compliance.
7. Vascular injury and pulmonary hypertension
Microvascular injury leads to:
- Microthrombi from fibrocellular proliferation, causing vascular obliteration
- Intrapulmonary shunting: blood flows past collapsed, unventilated alveoli → severe hypoxemia
- Increased dead space: microvascular occlusion reduces blood flow to ventilated lung regions → hypercapnia
- Pulmonary hypertension from vascular resistance rise
ENaC (epithelial sodium channels) and Na⁺/K⁺-ATPase on type II pneumocytes normally drive active fluid clearance from the alveolus. Hypoxia and injury impair these transporters, further impairing edema clearance.
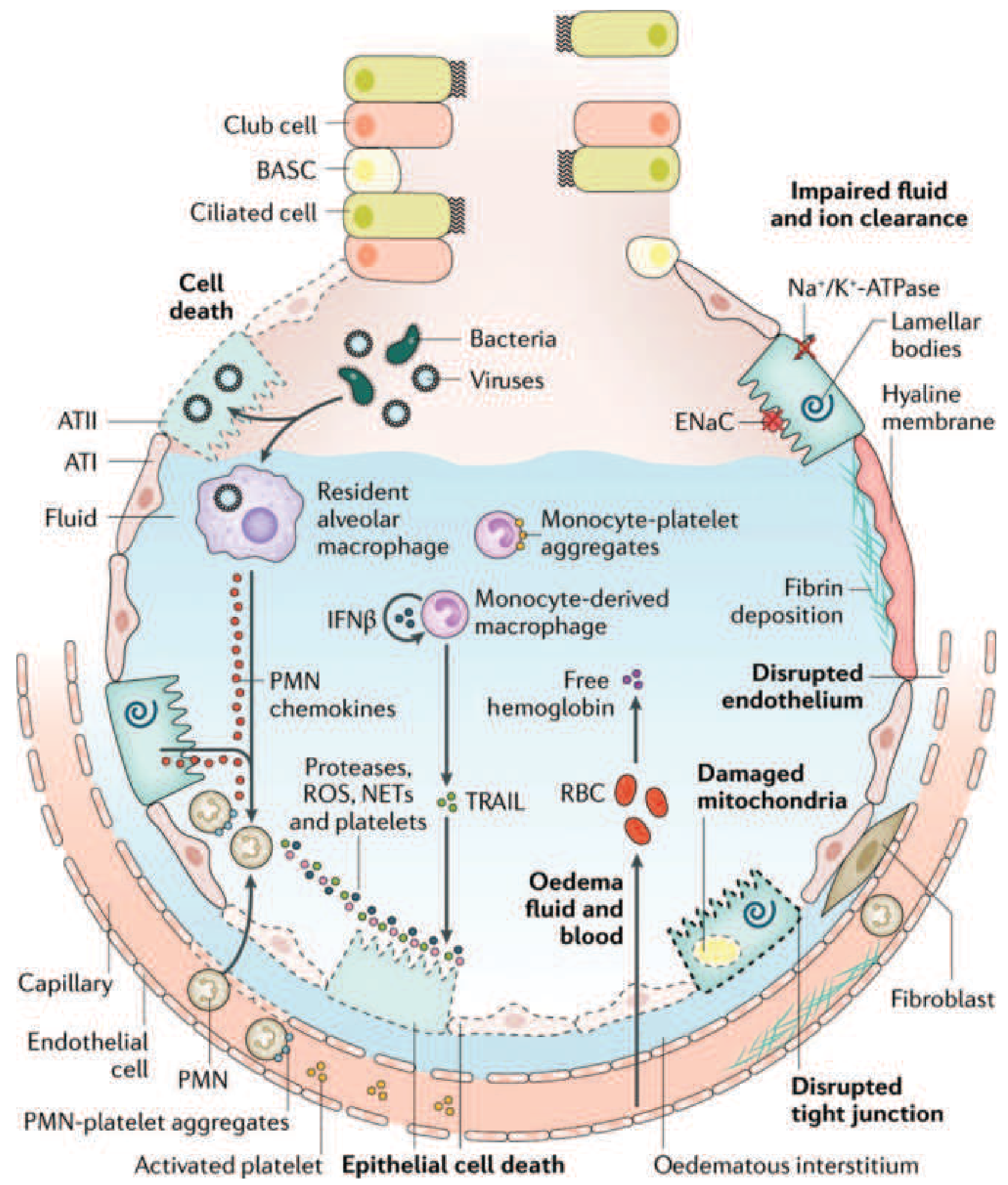
Phase 2: Proliferative Phase (Days 7-21)
This phase marks the beginning of lung repair:
- The inflammatory infiltrate shifts from neutrophil-predominant to lymphocyte-predominant
- Type II pneumocytes (which survived injury because they are more resistant than type I cells) proliferate along alveolar basement membranes. They synthesize new surfactant and differentiate into type I pneumocytes to restore the epithelial lining
- Alveolar exudates begin to organize (fibrous organization replaces the fluid exudate)
- Many patients recover during this phase; however, some develop progressive lung injury and early fibrotic changes
Phase 3: Fibrotic Phase (Week 3 onwards - in some patients)
If resolution fails, a fibrotic phase develops:
- Alveolar edema and inflammatory exudates convert to extensive alveolar-duct and interstitial fibrosis
- Marked disruption of acinar architecture leads to emphysema-like changes and large bullae
- Intimal fibroproliferation in pulmonary microcirculation causes progressive vascular occlusion and pulmonary hypertension
- Lung biopsy evidence of pulmonary fibrosis at any ARDS phase is independently associated with increased mortality
Physiologic consequences of this phase: reduced lung compliance, increased dead space, risk of pneumothorax, and oxygen dependence.
- Harrison's Principles of Internal Medicine, 22nd ed., Ch. 312
Summary of Key Pathophysiologic Consequences
| Mechanism | Consequence |
|---|---|
| Alveolar flooding + surfactant loss | Atelectasis, reduced compliance, diffuse bilateral opacities |
| Intrapulmonary shunting | Severe, refractory hypoxemia (hallmark) |
| Dead-space increase | Hypercapnia, elevated minute ventilation requirement |
| Microvascular occlusion | Pulmonary hypertension, right heart strain |
| Epithelial/endothelial destruction | Non-cardiogenic pulmonary edema (PCWP normal) |
| Impaired fluid clearance (ENaC/Na⁺K⁺-ATPase failure) | Persistent alveolar flooding despite intact hydrostatics |
Ventilator-Induced Lung Injury (VILI) - A Mechanistic Amplifier
Because ARDS is heterogeneous (dependent lung is collapsed/consolidated; nondependent lung is relatively spared), mechanical ventilation risks:
- Volutrauma/barotrauma: overdistension of "baby lung" - the small volume of compliant lung that is still available
- Atelectrauma: repetitive opening and closing of collapsed units at low lung volumes
These mechanisms can perpetuate and amplify the original alveolar injury, a major reason why low tidal volume ventilation (6 mL/kg predicted body weight) is the cornerstone of ARDS management.
- Goldman-Cecil Medicine, Ch. 91; Harrison's Principles of Internal Medicine, Ch. 312; Robbins & Kumar Basic Pathology
Key sources: Harrison's Principles of Internal Medicine 22nd ed. (Ch. 312), Goldman-Cecil Medicine (Ch. 90-91), Murray & Nadel's Textbook of Respiratory Medicine, Robbins & Kumar Basic Pathology.
This is a shared conversation. Sign in to Orris to start your own chat.