
PXL_20260518_061316499.jpg
Can you decode the first word written on marker like what it can be?
The image is rotated, so let me orient the text properly. The document is from Northwest General Hospital & Research Centre, and the handwritten notes (written sideways) read:
Looking at the first word at the top of the handwritten text — it appears to be "Dx" (short for Diagnosis), a very common medical abbreviation used by doctors when jotting clinical notes.
Here's my interpretation of all the visible handwritten text:
| Written | Likely meaning |
|---|---|
| Dx | Diagnosis |
| Cu. Celebro / Uis. | Possibly "Cu. Cerebro/Vascular" → Cerebrovascular (as in a CVA/stroke note) |
| K.N. | Likely initials (doctor or patient) |
| 163 | Could be a blood pressure reading (e.g., 163 mmHg systolic) or a ward/bed number |
Most probable first word: "Dx" = Diagnosis
This is a typical quick clinical notation on a hospital letterhead, likely a doctor's shorthand summary. The curved script before "Celebro" could also be "Cu." as an abbreviation or part of a name/term.
Hemophagocytic lymphohistiocytosis
Reading File
Finding Sources
Searching PubMed
"hemophagocytic lymphohistiocytosis" diagnosis treatment
Searching the Web
hemophagocytic lymphohistiocytosis HLH diagnosis criteria treatment 2024 2025
Reading File
Reading File
Reading File
Reading File
Searching Images
hemophagocytic lymphohistiocytosis bone marrow hemophagocytosis
Reading File
Loading Image
Reading File
Here is a comprehensive overview of Hemophagocytic Lymphohistiocytosis (HLH):
Hemophagocytic Lymphohistiocytosis (HLH)
Definition
HLH is a life-threatening hyperinflammatory syndrome driven by massive, uncontrolled activation of CD8+ cytotoxic T lymphocytes and macrophages, leading to a "cytokine storm" and progressive multiorgan failure. It exists in two major forms:
- Primary (genetic/familial): Mendelian-inherited defects in lymphocyte cytotoxicity
- Secondary (acquired): Triggered by infection, malignancy, or autoimmune disease
"HLH is a highly life-threatening condition of massive, uncontrolled inflammation... often leading to multiple organ failure and death if not treated promptly." — Harrison's Principles of Internal Medicine 22E (2025)
Pathophysiology
The core defect is failure of cytotoxic lymphocytes (CTLs and NK cells) to eliminate infected or abnormal cells, particularly those infected by viruses like EBV.
- Cytotoxic lymphocytes normally release perforin and granzymes → perforin forms pores in target cell membranes → granzymes induce apoptosis
- When this is defective → infected cells persist → continuous stimulation of CD8+ T cells
- CD8+ T cells switch to their cytokine-secretory mode → massive IFN-γ release → macrophage overactivation
- Macrophages begin phagocytosing blood cells (hemophagocytosis) and secrete IL-1β, IL-6, IL-18, TNF-α → cytokine storm → tissue destruction → multiorgan failure
Key cytokines: IFN-γ (pivotal), IL-1β, IL-6, IL-18, TNF-α, CXCL9
Classification
Primary HLH (Mendelian)
| Subtype | Gene | Protein |
|---|---|---|
| FHL2 | PRF1 | Perforin (30% of FHL) |
| FHL3 | UNC13D | Munc13-4 (30%) |
| FHL4 | STX11 | Syntaxin-11 (10%) |
| FHL5 | STXBP2 | Munc18-2 (20%) |
| XLP1 | SH2D1A | SAP adaptor protein |
| XLP2 | BIRC4 (XIAP) | Anti-apoptotic molecule |
| Griscelli syndrome type 2 | RAB27A | Rab27a |
| Chédiak-Higashi syndrome | LYST | Giant lysosomes in leukocytes |
Secondary HLH (Non-Mendelian)
- Infection-associated: EBV (most common trigger), CMV, HIV, tuberculosis, fungi, parasites
- Malignancy-associated: Especially peripheral T-cell lymphoma
- Autoimmune-associated (MAS-HLH): Systemic JIA, adult-onset Still's disease, SLE, vasculitis
- Transplant-associated
Clinical Features
| System | Findings |
|---|---|
| Constitutional | High fever (often presenting sign), sepsis-like picture |
| Reticuloendothelial | Hepatosplenomegaly, lymphadenopathy (~50%) |
| Hematologic | Bicytopenia/pancytopenia (thrombocytopenia most common) |
| Hepatic | Elevated transaminases, conjugated hyperbilirubinemia, elevated GGT |
| Coagulation | DIC, hypofibrinogenemia, severe bleeding |
| CNS | Seizures, decreased consciousness, meningism, cranial nerve palsies (~1/3 of FHL cases) |
| Skin | Petechiae, purpura, transient maculopapular rash |
| Metabolic | Hypertriglyceridemia, hyponatremia, hypoalbuminemia |
Diagnostic Criteria (HLH-2004)
≥5 of 8 criteria must be met:
- Fever
- Splenomegaly
- Bicytopenia — Hb <90 g/L, platelets <100×10⁹/L, neutrophils <1.0×10⁹/L
- Hypertriglyceridemia (fasting TG ≥3.0 mmol/L / ≥265 mg/dL) and/or hypofibrinogenemia (≤1.5 g/L)
- Hemophagocytosis on bone marrow/CSF/lymph node biopsy
- Ferritin ≥500 μg/L (markedly elevated ferritin is a key screening test)
- Low/absent NK cell activity
- Soluble IL-2 receptor (sCD25) ≥2400 U/mL
Note: The Histiocyte Society revised criteria in 2024 (hemophagocytosis is not always present early in disease; serial BM exams may be needed).
Additional Workup
- Flow cytometry: CD107a (LAMP-1) degranulation assay → defective exocytosis suggests UNC13D/STXBP2/STX11/RAB27A variants
- Intracellular perforin expression → absent suggests PRF1 variants
- Genetic sequencing — gold standard but weeks to result
- CSF analysis — elevated cells and protein in ~50%
- MRI brain — nonspecific but may show inflammatory changes
Diagnostic Images
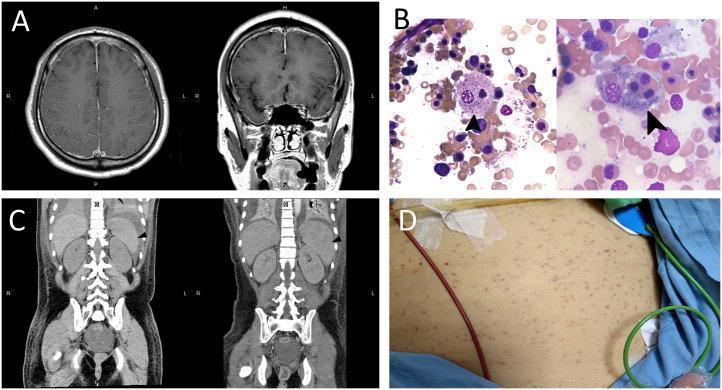
Multi-panel HLH findings: (A) MRI brain with bilateral dural thickening and vessel enhancement; (B) Bone marrow smears (×1000) — arrows show activated macrophages engulfing nucleated cells and platelets (hemophagocytosis); (C) CT abdomen showing spleen progression from normal to 12.7 cm; (D) Diffuse petechiae.
Treatment
Step 1 — Stop the Hyperinflammation (Pretransplant Therapy)
| Agent | Role |
|---|---|
| Dexamethasone | CNS-penetrant corticosteroid; backbone of HLH-94/HLH-2004 protocols |
| Etoposide | Selectively depletes activated T cells; suppresses cytokine production |
| Cyclosporin A | Reduces T-cell cytokine production |
| Emapalumab | Anti-IFN-γ monoclonal antibody (FDA-approved for primary/refractory HLH) |
| Ruxolitinib | JAK1/2 inhibitor; blocks multiple inflammatory cytokines |
| Alemtuzumab | Anti-CD52; targets lymphocytes |
Pre-HSCT survival with etoposide/alemtuzumab-based regimens: ~90% in experienced centers.
Step 2 — Curative Therapy
Allogeneic Hematopoietic Stem Cell Transplantation (HSCT) is the only curative treatment for primary HLH. Post-HSCT survival: 85–90%. Overall survival with treatment: ~80%.
- Also treat the triggering factor (e.g., antivirals for EBV, chemotherapy for underlying lymphoma)
- Secondary HLH: treat underlying cause + immunosuppression; HSCT reserved for refractory cases
Prognosis
- Untreated: Median survival 1–2 months (primary HLH); essentially fatal
- With treatment: ~50% overall survival across all forms; better outcomes at specialized centers
- Long-term complications: Neurologic sequelae in ~1/3 (psychomotor retardation, epilepsy, ADHD, ataxia), endocrinopathies (~10%)
- Secondary HLH in adults: Prognosis depends heavily on the underlying trigger
Sources: Harrison's Principles of Internal Medicine 22E (2025) | Robbins, Cotran & Kumar Pathologic Basis of Disease | Robbins & Kumar Basic Pathology
Recent systematic reviews (2024–2025) have highlighted specific HLH associations — tuberculosis-associated HLH (PMID 39581974), GATA2 deficiency-associated HLH (PMID 39497062), and IBD-associated HLH (PMID 40895566) — consistent with established pathophysiology.
This is a shared conversation. Sign in to Orris to start your own chat.