Ild
Reading File
Finding Sources
Searching PubMed
"interstitial lung disease" AND diagnosis
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Here is a thorough overview of Interstitial Lung Disease (ILD) based on Harrison's Principles of Internal Medicine 22E, Murray & Nadel's Textbook of Respiratory Medicine, The Washington Manual, and Fishman's Pulmonary Diseases.
Interstitial Lung Disease (ILD)
Definition
ILD (also called diffuse parenchymal lung disease) encompasses more than 200 heterogeneous conditions that affect the lung parenchyma with varying degrees of inflammation and fibrosis. The primary site of injury is the interstitium (the space between alveolar epithelium and capillary endothelium), though alveolar epithelial cells, endothelium, and airways are often involved.
Classification
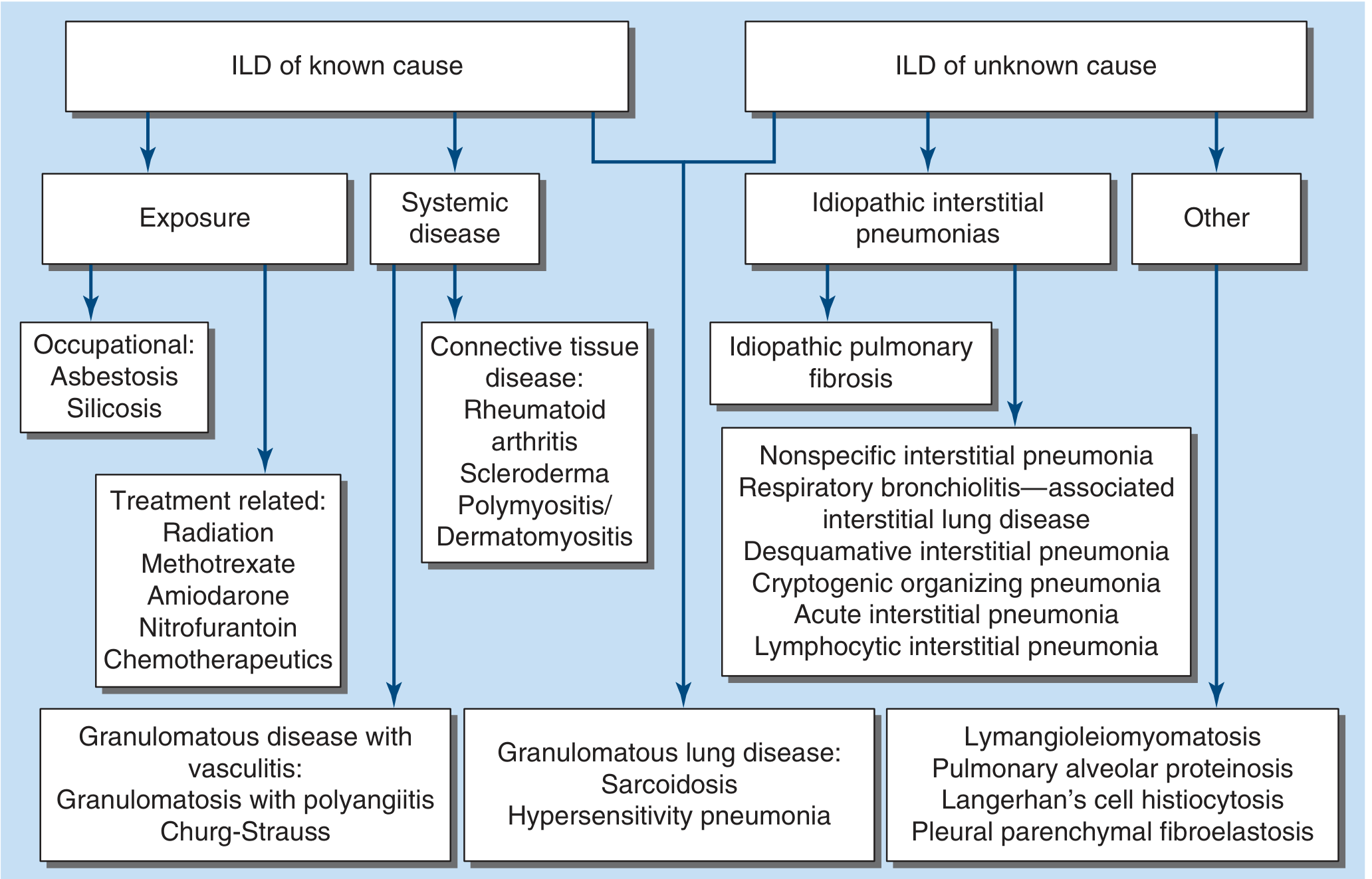
Figure 304-1: Classification of ILD - Harrison's Principles of Internal Medicine 22E
ILDs are divided broadly into known and unknown causes:
ILD of Known Cause
| Category | Examples |
|---|---|
| Occupational/Environmental exposure | Asbestosis, silicosis, coal worker's pneumoconiosis |
| Drug/treatment-related | Methotrexate, amiodarone, nitrofurantoin, radiation, chemotherapeutics |
| Connective tissue disease (CTD) | Rheumatoid arthritis, scleroderma (SSc), polymyositis/dermatomyositis, SLE |
| Granulomatous with vasculitis | GPA (Wegener's), Churg-Strauss (eosinophilic GPA) |
| Granulomatous lung disease | Sarcoidosis, hypersensitivity pneumonitis (HP) |
ILD of Unknown Cause (Idiopathic Interstitial Pneumonias - IIPs)
| Subtype | Notes |
|---|---|
| IPF (Idiopathic Pulmonary Fibrosis) | Most common and most lethal IIP |
| NSIP (Nonspecific Interstitial Pneumonia) | Often CTD-associated; better prognosis |
| RB-ILD (Respiratory Bronchiolitis-ILD) | Smoking-related |
| DIP (Desquamative Interstitial Pneumonia) | Smoking-related |
| COP (Cryptogenic Organizing Pneumonia) | Formerly BOOP; steroid-responsive |
| AIP (Acute Interstitial Pneumonia) | Rapidly progressive, Hamman-Rich syndrome |
| LIP (Lymphocytic Interstitial Pneumonia) | Associated with Sjogren's, HIV |
Other ILDs
- Lymphangioleiomyomatosis (LAM)
- Pulmonary alveolar proteinosis (PAP)
- Langerhans cell histiocytosis (LCH)
- Pleural parenchymal fibroelastosis (PPFE)
Clinical Presentation
| Symptom/Sign | Details |
|---|---|
| Dyspnea | Progressive exertional - the cardinal symptom |
| Dry cough | Persistent, nonproductive |
| Fine crackles | End-inspiratory, bibasilar ("Velcro rales") - especially in IPF |
| Digital clubbing | Common in IPF; less so in NSIP/RB-ILD |
| Cyanosis/cor pulmonale | Signs of advanced disease |
| Extrapulmonary features | Skin thickening (SSc), joint findings (RA), lymphadenopathy (sarcoidosis) |
Diagnostic Workup
A key tenet: no single test is diagnostic alone. Diagnosis requires integration of:
- Clinical history
- Lab studies
- PFTs
- Imaging
- Histopathology (if obtained)
1. History
- Smoking history (RB-ILD, DIP, IPF)
- Occupational/environmental exposures (asbestos, birds, molds for HP)
- Drug history
- Family history - familial IPF may account for up to 20% of cases; MUC5B promoter variant and TERT mutations are key genetic risk factors
2. Lab Studies
- Autoantibody panel for CTD: ANA, RF, anti-CCP, anti-Scl-70, anti-Jo-1, myositis-specific antibodies
- CBC, CMP (baseline for treatment monitoring)
3. Pulmonary Function Tests (PFTs)
- Restrictive pattern: reduced TLC, with proportionally reduced FEV1 and FVC (FEV1/FVC ratio normal or increased)
- Reduced DLCO: may precede volume loss; indicates impaired gas exchange
- Mixed pattern possible in sarcoidosis, HP, LAM
4. Chest Imaging
HRCT is the standard of care for initial ILD evaluation. CXR alone rarely leads to a specific diagnosis.
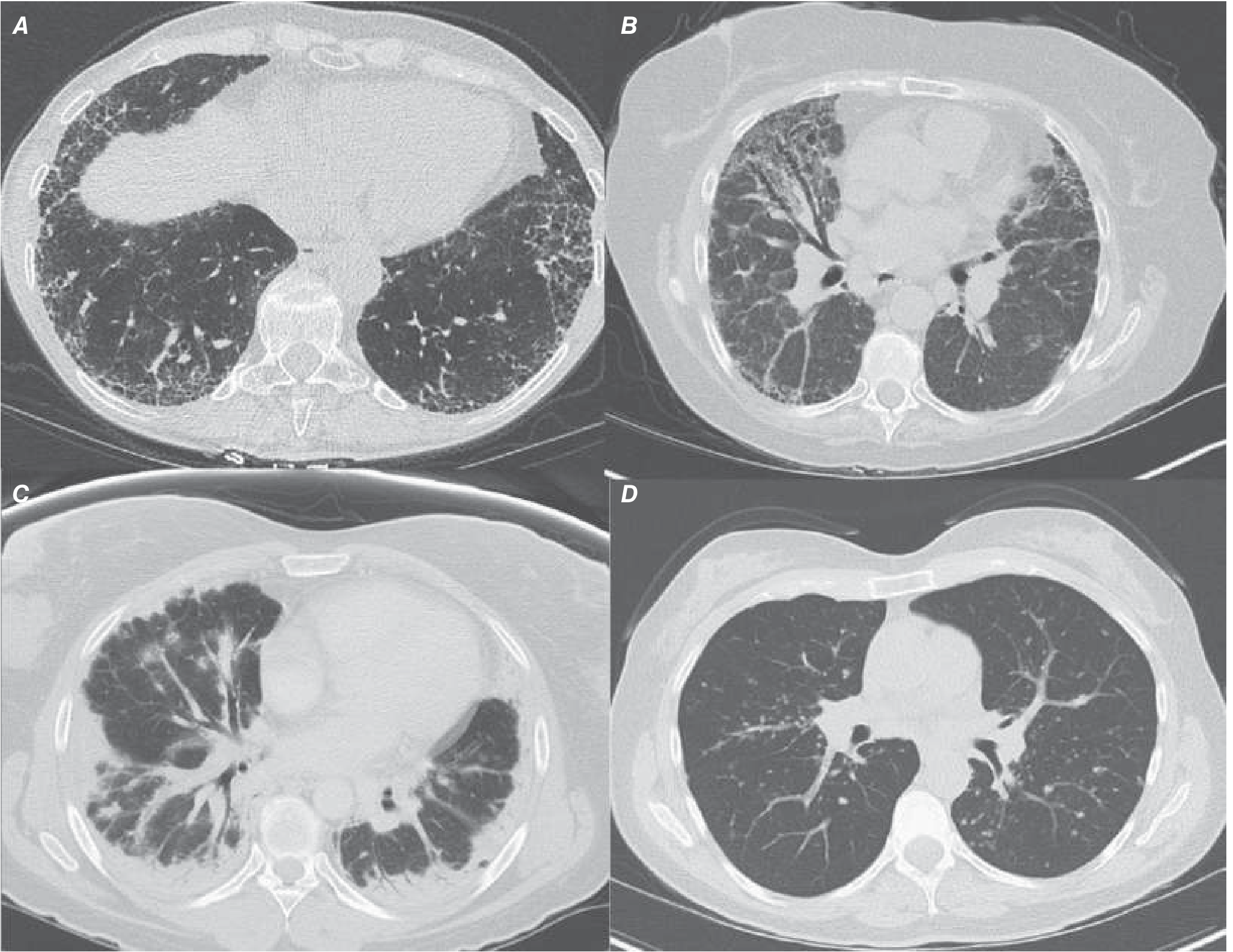
Figure 304-2: HRCT patterns - Harrison's 22E. A = IPF/UIP (posterior basilar reticular + honeycombing), B = NSIP (bilateral GGO + reticular, spares subpleura), C = COP (patchy consolidative opacities, reversed halo sign), D = Sarcoidosis (peribronchovascular nodules + hilar LAD)
| Pattern | Typical ILD |
|---|---|
| Bilateral basilar subpleural reticular + honeycombing + traction bronchiectasis | UIP/IPF |
| Bilateral symmetric ground-glass + reticular, subpleural sparing | NSIP |
| Patchy subpleural consolidation, reversed halo (atoll sign) | COP |
| Centrilobular ground-glass nodules | RB-ILD |
| Peribronchovascular nodules + hilar LAD | Sarcoidosis |
| Upper lobe predominance + air trapping | HP |
5. Bronchoscopy & Biopsy
- BAL: useful for HP (lymphocytosis), DIP/RB-ILD (smoker's macrophages), PAP (milky fluid), infection exclusion
- Transbronchial biopsy: sufficient for sarcoidosis; too small for most other ILDs
- Surgical lung biopsy (VATS): gold standard for definitive histopathologic diagnosis when HRCT is non-diagnostic; risks must be weighed
Key Histopathologic Patterns
| Pattern | Associated Disease |
|---|---|
| UIP (Usual Interstitial Pneumonia) | IPF, CTD-ILD (RA, SSc) |
| NSIP | CTD-ILD, idiopathic NSIP |
| OP (Organizing Pneumonia) | COP, drug-induced, infection |
| DIP | Smoking-related ILD |
| Respiratory bronchiolitis | RB-ILD |
| Noncaseating granulomas | Sarcoidosis, HP |
Management
Treatment is disease-specific:
| ILD Type | Treatment |
|---|---|
| IPF | Pirfenidone or Nintedanib (antifibrotics); lung transplant for eligible patients |
| CTD-ILD (UIP/NSIP/COP pattern) | Corticosteroids + immunosuppressives (cyclophosphamide, azathioprine, mycophenolate, rituximab) |
| Sarcoidosis | Corticosteroids; methotrexate, azathioprine, or infliximab for refractory cases |
| HP | Antigen avoidance; corticosteroids; antifibrotic therapy if progressive fibrosis develops |
| Drug-induced ILD | Discontinue culprit drug; corticosteroids |
| RB-ILD / DIP | Smoking cessation; corticosteroids of limited benefit |
| COP | Corticosteroids (highly responsive) |
Antifibrotics in Detail
- Pirfenidone (oral): inhibits IL-1β, IL-6, TNF-α, PDGF, TGF-β; slows FVC decline in IPF. Side effects: skin rash (30%), photosensitivity, nausea, diarrhea. Monitor LFTs.
- Nintedanib (oral tyrosine kinase inhibitor against FGFR, VEGFR, PDGFR): slows FVC decline in IPF and now approved for progressive pulmonary fibrosis of other types. Side effects: diarrhea (62%), nausea, vomiting. Monitor LFTs.
- Important: the triple therapy of N-acetylcysteine + azathioprine + prednisone is contraindicated in IPF due to increased mortality.
Supportive Care
- Supplemental O2 when indicated
- Pulmonary rehabilitation (improves 6-minute walk distance and quality of life)
- Lung transplantation for eligible end-stage patients
Prognosis (Selected)
| ILD | Approximate Mortality |
|---|---|
| IPF | 50% at 3-5 years |
| NSIP | 18% at 5 years |
| RB-ILD | 25% at 7 years |
| SSc-ILD | 20-30% at 10 years |
| Sarcoidosis | Generally low, variable |
Recent Evidence (2024-2025)
- A meta-analysis in Chest (2024) (PMID 38821182) examined pulmonary hypertension in ILD, finding it to be a significant complication associated with worsened outcomes.
- ERS guidelines for pulmonary alveolar proteinosis (PMID 39147411) were updated in 2024.
Sources: Harrison's Principles of Internal Medicine 22E (2025), p. 2303-2306; The Washington Manual of Medical Therapeutics; Murray & Nadel's Textbook of Respiratory Medicine; Fishman's Pulmonary Diseases and Disorders
This is a shared conversation. Sign in to Orris to start your own chat.