Teach me like neonatologist, paeditrcian in depth topic - Плесента. Formation of плесента. Functions of placenta, placental abnormalities.
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
Reading File
I now have comprehensive content from multiple authoritative textbooks. Let me compile the full in-depth teaching response.
The Placenta (Плацента) - A Complete Neonatologist/Paediatrician's Perspective
Sources Used
- The Developing Human: Clinically Oriented Embryology (Moore & Persaud)
- Histology: A Text and Atlas (Ross & Pawlina)
- Robbins, Cotran & Kumar: Pathologic Basis of Disease
- Creasy & Resnik's Maternal-Fetal Medicine
1. WHAT IS THE PLACENTA?
The placenta is a fetomaternal organ - it has two distinct components:
- Fetal part - formed by the chorion (outermost fetal membrane derived from the chorionic sac)
- Maternal part - formed by the decidua basalis (the endometrium directly beneath the implanted embryo)
At term, the placenta is disc-shaped, 15-20 cm in diameter, 2-3 cm thick, and weighs 500-600 g - approximately one-sixth the weight of the fetus.
2. FORMATION OF THE PLACENTA
2.1 The Decidua - First Step
When the blastocyst implants, the endometrium transforms into the decidua. Three regions are named by their relationship to the implantation site:
| Region | Location | Role |
|---|---|---|
| Decidua basalis | Deep to the conceptus | Forms the maternal part of the placenta |
| Decidua capsularis | Superficial, overlying the conceptus | Disappears as pregnancy advances |
| Decidua parietalis | All remaining endometrium | Lines the rest of the uterine cavity |
In response to rising progesterone, connective tissue cells enlarge to form decidual cells packed with glycogen and lipids. The decidual reaction (cellular and vascular changes at implantation) provides nutrition for the early embryo.
2.2 Trophoblast Invasion and the Lacunar Stage (Week 2)
Around day 9 after fertilization, the syncytiotrophoblast erodes maternal capillaries forming trophoblastic lacunae (blood-filled spaces). Maternal sinusoids anastomose with these lacunae, establishing a primitive uteroplacental circulation - driven by arteriovenous pressure differentials. Numerous pinocytotic vesicles in the syncytiotrophoblast allow transfer of nutrients to the embryo.
This is the era of hemotrophic nutrition (via maternal blood) replacing the earlier histiotrophic nutrition (via decidual cell breakdown products).
2.3 Chorionic Villi Development - The Core of Placentation
This is the most important structural event in placental formation:
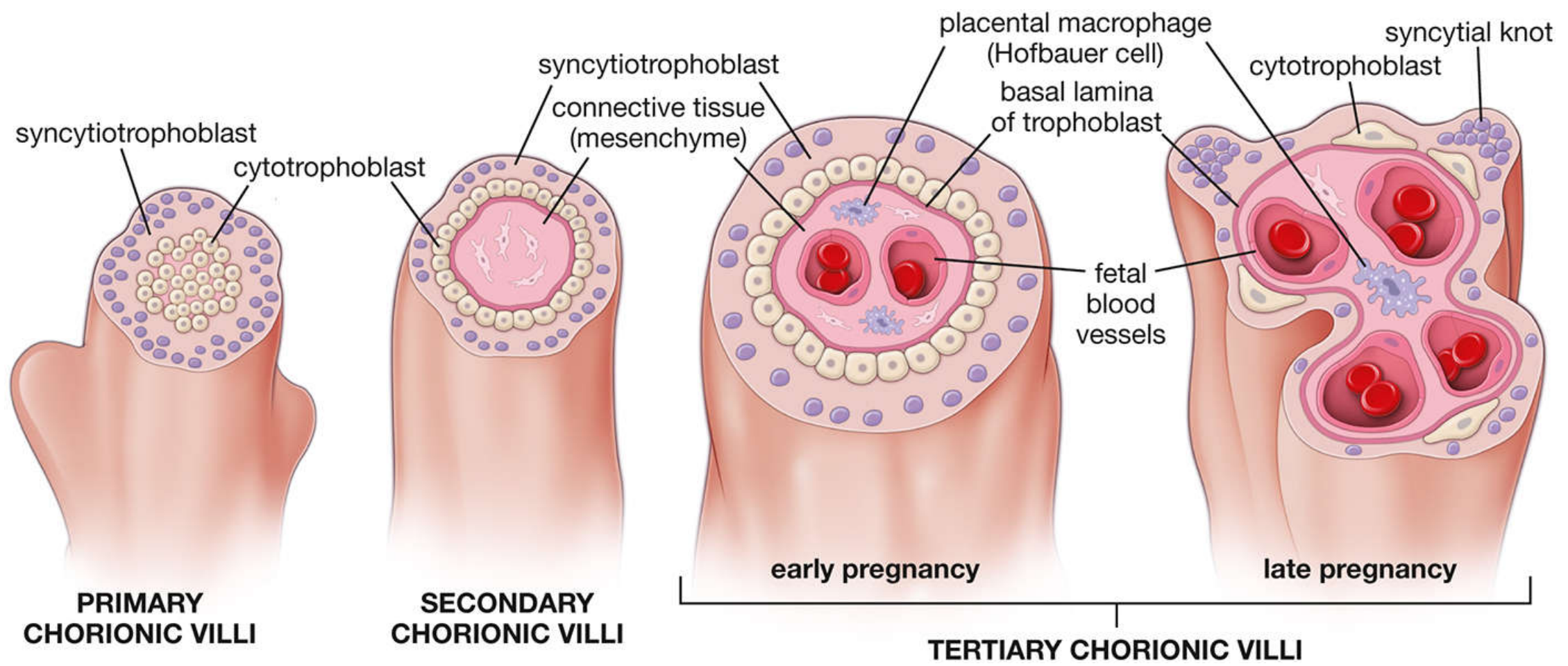
Primary villi (end of Week 2)
- Syncytiotrophoblast + cytotrophoblast form finger-like projections into the decidua
- No mesenchyme or vessels yet
Secondary villi (early Week 3)
- Extraembryonic mesenchyme (connective tissue) grows into the villi core
- Still surrounded by cytotrophoblast and syncytiotrophoblast
Tertiary villi (late Week 3)
- Blood vessels and supportive cells (including Hofbauer cells - placental macrophages) differentiate within the mesenchymal core
- Fetal blood vessels now form; this is functional vasculogenesis
Maturation during pregnancy:
- Early pregnancy: Large, edematous villi, few vessels, many connective tissue cells, thick syncytiotrophoblast + continuous cytotrophoblast layer
- Late pregnancy: Cytotrophoblast becomes discontinuous, syncytiotrophoblast nuclei aggregate into syncytial knots, more fetal blood vessels, less cellular stroma
2.4 The Cytotrophoblastic Shell and EVT Invasion
A key event: cytotrophoblast cells break out from anchoring villi and form:
- Cytotrophoblastic shell - continuous cell layer separating decidua from maternal blood sinuses
- Interstitial trophoblast - cells dispersed within decidua
- Endovascular trophoblast (EVT) - adopts an endothelial phenotype, invades and remodels the spiral arteries, replacing their tunica media and endothelium, converting them into wide, low-resistance, high-flow channels
This arterial remodeling is physiologically critical: it ensures stable, high-flow perfusion of the intervillous space regardless of maternal vasoactive molecules. Inadequate EVT invasion → poor perfusion → preeclampsia and fetal growth restriction (FGR).
2.5 Chorion Frondosum vs. Chorion Laeve
As the chorionic sac grows:
- Chorion frondosum ("leafy chorion"): villi adjacent to the decidua basalis - proliferate vigorously to form the fetal placenta
- Chorion laeve ("smooth chorion"): villi on the side facing the decidua capsularis - degenerate and disappear as blood supply diminishes
The amniotic sac expands rapidly, fusing the amnion with the chorion laeve to form the amniochorionic membrane - the bag of waters that ruptures during labor.
2.6 Final Architecture at Full Term
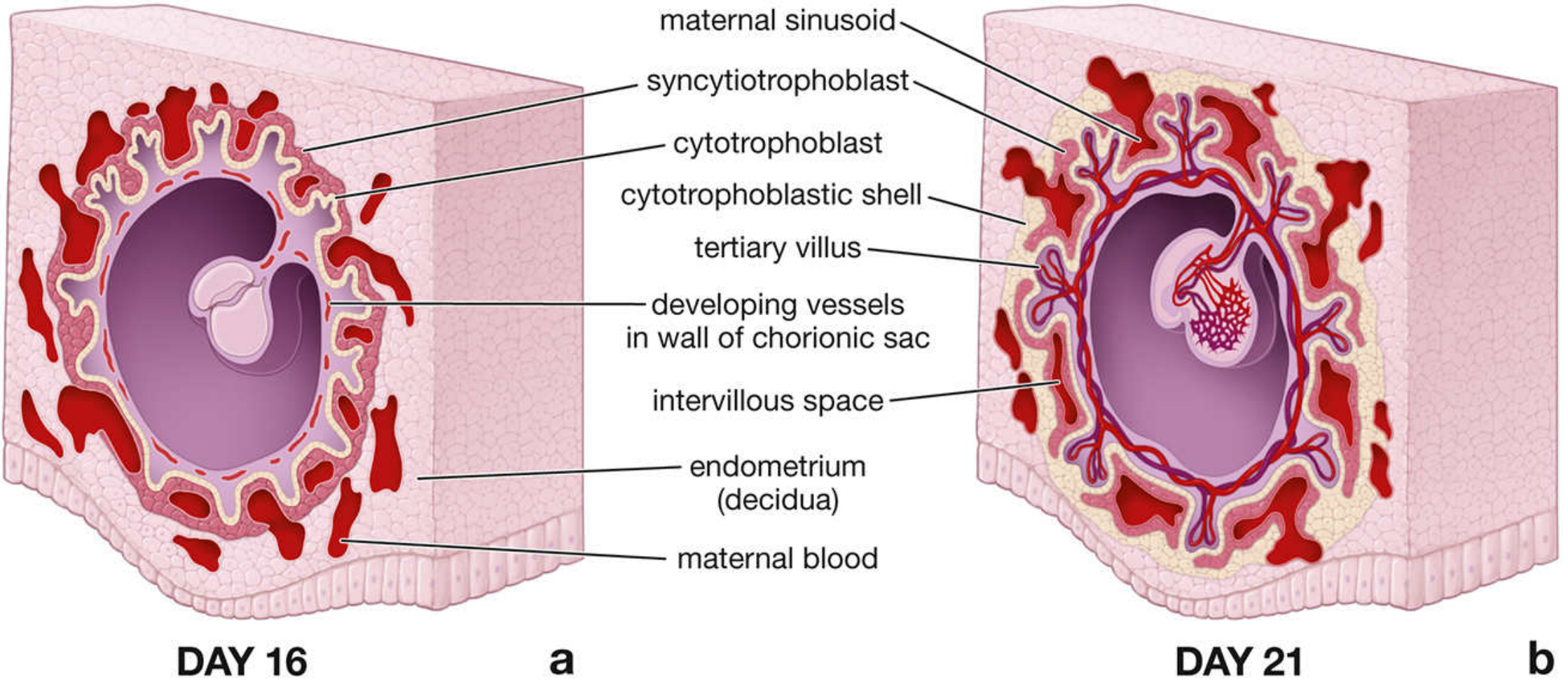
The definitive placenta is organized into 15-30 cotyledons, each consisting of a main stem villus (truncus chorii) with many branch villi. Cotyledons are separated by placental septa projecting from the basal plate. The intervillous space is bathed by maternal arterial blood (600 mL/min at term) via 80-100 spiral arteries.
3. PLACENTAL CIRCULATION
3.1 Fetal Circulation
- Deoxygenated blood: fetus → umbilical arteries (2) → divide at placental surface → chorionic arteries → chorionic villi capillaries
- Oxygenated blood: thin-walled veins → converge → umbilical vein (1) → fetus
- No intermingling of fetal and maternal blood under normal conditions (microscopic breaks can allow feto-maternal hemorrhage)
3.2 Maternal Circulation
Maternal blood enters the intervillous space through spiral arteries as high-pressure jets, circulates around the villi, then returns via endometrial veins. The entire intervillous space turns over approximately 3-4 times per minute.
3.3 The Placental Membrane (Placental Barrier)
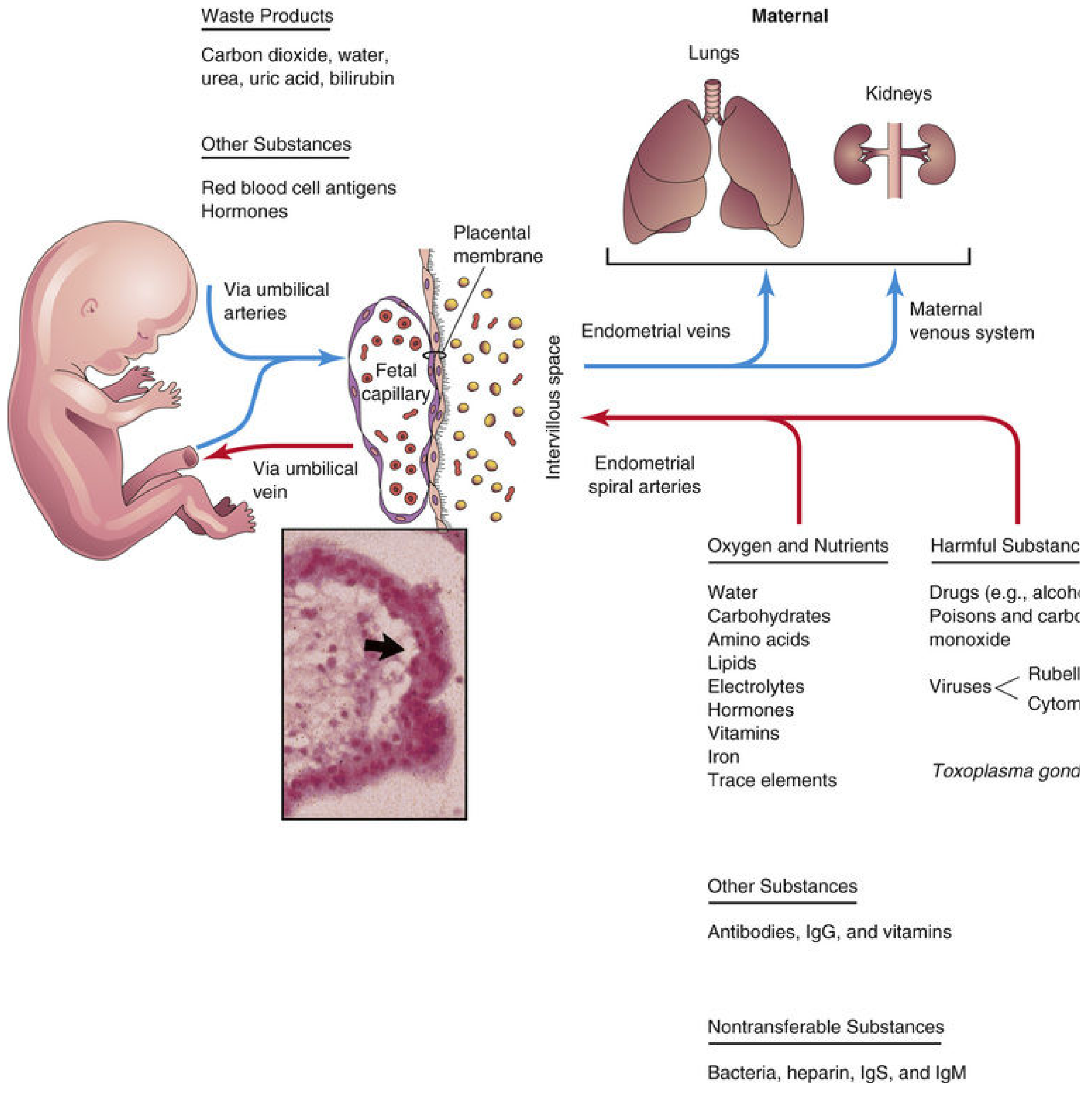
Four layers separate fetal from maternal blood (early pregnancy):
- Syncytiotrophoblast
- Cytotrophoblast
- Connective tissue (mesenchyme)
- Fetal capillary endothelium
In late pregnancy, cytotrophoblast thins out, reducing this to 2-3 layers, which increases exchange efficiency. This thinning is critical for late-gestation gas and nutrient transfer.
4. FUNCTIONS OF THE PLACENTA
4.1 Respiratory Function (Gas Exchange)
- O₂, CO₂, carbon monoxide - all by simple diffusion
- O₂ transport is flow-limited, not diffusion-limited → fetal hypoxia results from reduced uterine or fetal blood flow, not from diffusion failure
- Fetal hemoglobin (HbF) has higher O₂ affinity than adult HbA → facilitates O₂ loading from maternal blood
- Placental efficiency for gas exchange approaches that of the lungs
4.2 Nutritional Function
| Substance | Transport Mechanism |
|---|---|
| Water | Simple diffusion (increasing amounts with advancing gestational age) |
| Glucose | Facilitated diffusion via GLUT-1 (insulin-independent) |
| Amino acids | Active transport (fetal levels exceed maternal levels) |
| Free fatty acids | Simple diffusion (long-chain PUFAs in highest amounts) |
| Vitamins | Cross by diffusion |
| Iron | Active transport via transferrin receptors |
| Cholesterol, triglycerides, phospholipids | Transferred from maternal side |
Clinical note: Glucose is the primary fetal energy substrate. Any impairment of GLUT-1 or reduced maternal glucose availability directly affects fetal growth.
4.3 Excretory Function
Fetal waste products transferred to maternal blood for elimination:
- CO₂, water, urea, uric acid, bilirubin → maternal kidneys and lungs excrete them
- Bilirubin in particular is important: the fetus cannot conjugate bilirubin efficiently, so unconjugated bilirubin crosses to the mother for hepatic clearance. This is why severe hyperbilirubinemia does not accumulate prenatally.
4.4 Protective (Barrier) Function
Substances that DO NOT cross:
- Bacteria (in most cases)
- Heparin
- IgS (large molecules)
- IgM (does not cross the placenta - important clinically: elevated IgM in cord blood = congenital infection)
Substances that DO cross:
- IgG - actively transported (neonatal passive immunity; peaks at term)
- Drugs (most cross by diffusion - clinically vital: opioids, sedatives, alcohol, cocaine)
- Viruses (CMV, rubella, herpes, varicella, HIV)
- Toxoplasma gondii, Treponema pallidum
Clinical pearl: IgG transfer is the basis for neonatal passive immunity. Premature infants have low IgG (transfer is highest in the 3rd trimester, especially after 32 weeks), making them more susceptible to infection.
4.5 Endocrine/Hormonal Function
The syncytiotrophoblast is a hormonally active tissue producing:
Protein Hormones:
| Hormone | Timing | Role |
|---|---|---|
| hCG (human chorionic gonadotropin) | Secreted from week 2; peaks at week 8 | Maintains corpus luteum, prevents menstruation; basis of pregnancy tests |
| hCS / hPL (human placental lactogen) | Rises through pregnancy | Decreases maternal glucose utilization → spares glucose for fetus; increases maternal FFA availability |
| hCT (human chorionic thyrotropin) | Through pregnancy | Functions like TSH |
Steroid Hormones:
| Hormone | Substrate | Role |
|---|---|---|
| Progesterone | Maternal cholesterol | Maintains uterine quiescence; prevents abortion |
| Estrogens | Fetal adrenal androgens (DHEAS) | Uterine growth, breast development |
Clinical note: After the 1st trimester, the placenta takes over progesterone production from the corpus luteum. The fetal-placental-maternal unit (fetoplacental unit) cooperates in estrogen biosynthesis: the fetal adrenal provides the androgen substrate that the placenta aromatizes to estrogens.
4.6 Immunological Function - The Allograft Paradox
The placenta is an allograft - it carries paternal antigens yet is not rejected by the mother. Several mechanisms protect it:
- Classical HLA absence: Syncytiotrophoblast does NOT express classical HLA-A or HLA-B antigens that would trigger T-cell recognition
- Non-classical HLA expression: Expresses HLA-G and HLA-E, which suppress NK-cell killing
- Complement regulation: Expresses complement-regulatory proteins (CD46/membrane cofactor protein) that block C3 activation
- IDO (indoleamine 2,3-dioxygenase): In trophoblast cells, depletes tryptophan locally, suppressing T-cell responses
- Epigenetic silencing: In decidual stromal cells, T-cell-attracting chemokines are epigenetically silenced (repressive histone marks)
- Apoptosis induction: Apoptosis-inducing ligands on trophoblast delete activated maternal leukocytes
Clinical relevance: Failure of these mechanisms may contribute to recurrent miscarriage and preeclampsia.
5. PLACENTAL ABNORMALITIES
5.1 Abnormalities of Implantation (Placenta Accreta Spectrum)
When the decidua is absent (focally or completely), villous tissue contacts or invades myometrium directly:
| Term | Definition | Depth |
|---|---|---|
| Placenta accreta | Villi abnormally adhere to myometrium (no decidua) | ~0.2% of pregnancies |
| Placenta increta | Villi invade into myometrium | More severe |
| Placenta percreta | Villi penetrate through the full myometrial wall to serosa or adjacent organs (bladder) | Most severe |
Predisposing factors:
- Placenta previa (up to 60% of accreta cases)
- Previous cesarean section or uterine surgery (endometrial scarring)
- Advanced maternal age, multiparity
Presentation: 3rd trimester bleeding, failure of placental separation after delivery → life-threatening postpartum hemorrhage
Management: Planned preterm cesarean hysterectomy, leaving placenta in situ if hysterectomy is not performed (conservative management)
5.2 Placenta Previa
- Placenta implants over or adjacent to the internal cervical os
- Types: complete (covering os entirely), partial, marginal, low-lying
- Presentation: Painless, bright-red vaginal bleeding in the 3rd trimester (classically at 30-32 weeks)
- Diagnosis: Ultrasound (transvaginal is most accurate)
- Management: Complete placenta previa → cesarean delivery mandatory (vaginal delivery risks catastrophic hemorrhage)
5.3 Placental Abruption (Abruptio Placentae)
- Premature separation of a normally implanted placenta from the uterine wall
- Presentation: Painful dark vaginal bleeding, rigid ("board-like") uterus, fetal distress
- Causes: Hypertension (most common), trauma, cocaine use, thrombophilias, sudden uterine decompression (e.g., after PPROM)
- Neonatal consequences: Fetal hypoxia, stillbirth, FGR, neonatal coagulopathy
5.4 Placental Insufficiency and Fetal Growth Restriction (FGR)
During the third trimester, vigorous fetal growth places heavy demands on uteroplacental blood supply. Placental insufficiency is a major cause of FGR:
Causes of uteroplacental insufficiency:
- Single umbilical artery and abnormal cord insertion
- Placental abruption
- Placenta previa
- Placental thrombosis and infarction
- Chronic villitis of unknown etiology (CVUE)
- Placental infection
- Multiple gestations
Type of FGR: Placental causes typically produce asymmetric (head-sparing) FGR - body/visceral growth is restricted while brain growth is relatively preserved ("brain-sparing" effect via blood flow redistribution through ductus venosus and foramen ovale).
Neonatal consequences of FGR (SGA infant):
- Perinatal asphyxia, hypoglycemia, polycythemia, hypothermia
- Increased risk of cerebral dysfunction, learning disability, hearing/visual impairment
- Long-term: metabolic syndrome, cardiovascular disease in adult life (Barker hypothesis)
5.5 Placental Infections
Route 1 - Ascending infection (most common):
- Bacteria ascend from the birth canal
- Cause chorioamnionitis: amniotic fluid becomes cloudy/purulent; chorion-amnion shows maternal neutrophil infiltrate
- Premature rupture of membranes (PROM) and preterm labor
- Fetal inflammatory response: Vasculitis of umbilical vessels and fetal chorionic plate vessels → intense fetal inflammatory response is associated with neonatal morbidity and cerebral palsy
Route 2 - Hematogenous (transplacental):
- From maternal sepsis, viremia, or parasitemia
- TORCH organisms (Toxoplasmosis, Others [syphilis, TB, listeriosis], Rubella, CMV, Herpes): cause chronic villitis (chronic inflammatory infiltrate in chorionic villi)
- Associated with FGR
- CMV causes acute villitis histologically
5.6 Preeclampsia and Eclampsia (Placental Origin)
- Caused by inadequate EVT invasion → poorly remodeled spiral arteries → ischemic placenta releases anti-angiogenic factors (especially sFlt-1) and reduced VEGF and PlGF
- sFlt-1 causes widespread maternal endothelial dysfunction
- Manifestation: New onset hypertension + proteinuria after 20 weeks + systemic features
- HELLP syndrome (in ~10% of preeclampsia): Hemolysis + Elevated Liver enzymes + Low Platelets
- Eclampsia: Seizures - the most severe manifestation
- Responsible for >50,000 maternal deaths yearly worldwide
- More common in primigravidas, Black/Hispanic patients, multifetal pregnancies
5.7 Gestational Trophoblastic Disease
- Represents the opposite extreme of EVT invasion - excessive invasion
- Ranges from hydatidiform mole (benign) → invasive mole → choriocarcinoma (malignant)
- hCG is markedly elevated and serves as a tumor marker
5.8 Shape and Structural Abnormalities
| Abnormality | Description | Significance |
|---|---|---|
| Membranous placenta | Villi persist on entire chorionic sac surface; thin but large | Increased hemorrhage risk |
| Accessory (succenturiate) lobe | Extra lobe separated from main placenta | Risk: retained lobe after delivery |
| Bidiscoid placenta | Two lobes | Usually benign |
| Battledore placenta | Cord inserts at placental margin | Cord compression risk |
| Velamentous cord insertion | Umbilical vessels travel within fetal membranes before reaching placenta | Risk of vessel rupture and fetal exsanguination (vasa previa) |
| Circumvallate placenta | Chorionic plate is smaller than basal plate; membrane fold at edge | Associated with abruption, preterm labor |
5.9 Single Umbilical Artery (SUA)
- Normal cord has 2 arteries + 1 vein
- Single umbilical artery found in ~1% of deliveries
- Associated with congenital anomalies (cardiac, renal, chromosomal) in ~25-50% of isolated SUA
- Warrants fetal echocardiography and detailed anomaly scan
6. THE PLACENTA AND LONG-TERM DISEASE (The Barker Hypothesis)
The placenta links fetal programming to adult disease. Placental insufficiency causing FGR is associated with:
- Cardiovascular disease in adult life (hypertension, coronary artery disease)
- Type 2 diabetes mellitus
- Metabolic syndrome
This forms the basis of the Developmental Origins of Health and Disease (DOHaD) hypothesis - the intrauterine environment programs lifelong metabolic set-points.
CLINICAL SUMMARY TABLE FOR NEONATOLOGISTS
| Placental Issue | Neonatal Impact | Immediate Action |
|---|---|---|
| Abruption | Asphyxia, anemia, coagulopathy | Cord blood gas, CBC, crossmatch |
| Placenta previa (C/S) | Anemia, iatrogenic prematurity | Monitor for RDS, anemia |
| Placental insufficiency / FGR | Hypoglycemia, polycythemia, hypothermia | Dextrose infusion, hematocrit |
| Chorioamnionitis | Early-onset sepsis, pneumonia, NEC | Cultures, empiric antibiotics |
| TORCH infections | IUGR, hepatosplenomegaly, CNS injury | Specific serology, ophthalmology |
| Single umbilical artery | Congenital anomalies | Echo, renal US, chromosomes |
| Preeclampsia (premature birth) | Prematurity complications, thrombocytopenia | Platelet count, glucose |
Key References:
- The Developing Human: Clinically Oriented Embryology (Moore & Persaud), Chapter 7
- Histology: A Text and Atlas (Ross & Pawlina), Chapter 23
- Robbins Pathologic Basis of Disease, Chapters 22 & 10
- Creasy & Resnik's Maternal-Fetal Medicine, Chapter on Placenta
Teach me like neonatologist, paeditrcian in depth topic - Tetralogy including caustitive factors for teratogenicity, mode of action of teratogens, critical periods
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Loading Image
I now have all the content needed. Let me compile the comprehensive teaching response.
Teratology - Complete Neonatologist/Paediatrician's Teaching Guide
Sources Used
- The Developing Human: Clinically Oriented Embryology (Moore & Persaud) - primary source
- Robbins & Kumar Basic Pathology
- Rosen's Emergency Medicine
- The Harriet Lane Handbook (Johns Hopkins, 23rd ed.)
PART 1: TERATOLOGY - FOUNDATIONS
What is a Teratogen?
A teratogen is any agent that can produce a birth defect (congenital anomaly) or increase the incidence of a defect in the population. Environmental factors (drugs, infections, chemicals, radiation) cause 7-10% of all birth defects.
Birth defect causes overall:
- Genetic/chromosomal: ~30%
- Multifactorial (gene + environment): ~25%
- Environmental (teratogens): 7-10%
- Unknown: ~50% (most common category)
PART 2: CAUSATIVE FACTORS FOR TERATOGENICITY
2.1 Categories of Teratogens
A. Drugs and Chemicals
The teratogenicity of drugs varies enormously. Some (e.g., thalidomide) cause devastating defects at precise windows of exposure; others cause subtle cognitive or functional deficits with prolonged use throughout gestation. Between 40-90% of women take at least one non-prescription drug during pregnancy, yet less than 2% of birth defects are directly attributable to drugs and chemicals.
| Drug / Chemical | Critical Window | Defects Produced |
|---|---|---|
| Thalidomide | 20-36 days post-fertilization | Phocomelia/amelia (limb reduction), ear anomalies, CHD, gut atresia |
| Alcohol (ethanol) | Any trimester; no safe amount | FASD: microcephaly, short palpebral fissures, smooth philtrum, thin upper lip, cognitive deficiency, CHD |
| Isotretinoin (retinoic acid) | Weeks 3-5 | Microtia/anotia, depressed nasal bridge, CHD, brain malformations, intellectual disability |
| Phenytoin | Organogenesis | Fetal hydantoin syndrome: growth deficiency, hypertelorism, flat nasal bridge, cleft lip/palate, digital hypoplasia |
| Valproate | Organogenesis | High forehead, broad nasal bridge, cardiac defects, neural tube defects (NTD), spina bifida |
| Warfarin | Weeks 6-12 | Nasal hypoplasia, stippled epiphyses, hypoplastic phalanges, brain malformations |
| Tetracyclines | Week 14 to 8 years postnatal | Tooth enamel hypoplasia, brown/yellow discoloration, impaired long bone growth |
| Streptomycin | Any | CN VIII damage - deafness and vestibular damage |
| ACE inhibitors | 2nd/3rd trimester | Oligohydramnios, renal tubular dysgenesis, skull ossification defects |
| Methotrexate | Organogenesis | Microcephaly, micrognathia, low-set ears, growth restriction |
| DES (diethylstilbestrol) | Prenatal | Vaginal adenosis, clear cell adenocarcinoma (in daughters in 2nd decade); epididymal cysts (in sons) |
| Cocaine | Any | IUGR, developmental delay, occasional anomalies |
| Progestins (ethisterone, norethisterone) | Critical period | Cardiovascular defects, hypospadias, VACTERL association |
Fetal Alcohol Spectrum Disorder (FASD) - Key Points:
- Maternal alcohol abuse is the most common preventable cause of cognitive deficiency
- FAS prevalence: 1-2/1000 live births; FASD prevalence: ≥1%
- Mechanism: ethanol → acetaldehyde → disrupts cell adhesion, cell migration, apoptosis, and neurotrophic factor signaling
- Sentinel facial features: short palpebral fissures, smooth philtrum, thin vermilion border (upper lip), flat nasal bridge, small chin
- No safe amount of alcohol during pregnancy - the brain is susceptible throughout gestation
B. Cigarette Smoking
- Well-established cause of IUGR (intrauterine growth restriction)
- Low birth weight (<2000g) is the chief predictor of infant death
- Among heavy smokers, premature delivery is twice as frequent
- Mechanism: Nicotine constricts uterine blood vessels → reduced uteroplacental blood flow → ↓O₂ and nutrient delivery. Carboxyhemoglobin (from CO) impairs O₂ transport
- Modest increased risk of conotruncal and atrioventricular septal defects
- Associated with smaller brain volumes in preterm infants
C. Infectious Agents (Biological Teratogens)
| Organism | Route | Key Neonatal Features | Most Sensitive Period |
|---|---|---|---|
| Rubella | Transplacental | Cataracts, CHD (PDA, PS), deafness (classic triad) + cognitive deficiency, glaucoma, microphthalmia | Weeks 1-5 (80% risk); weeks 6-12 (~15%) |
| CMV | Transplacental (most common congenital viral infection - 1% neonates) | Developmental delay, IUGR, microphthalmia, chorioretinitis, microcephaly, cerebral calcification (periventricular), deafness, cerebral palsy, hepatosplenomegaly | Any (most severe if 1st trimester) |
| Toxoplasma gondii | Transplacental | Hydrocephalus, intracranial calcification, chorioretinitis (classic triad), IUGR | Any |
| Treponema pallidum | Transplacental | Bone destruction, CNS damage, skin lesions, saddle nose | After week 16 (treponema) |
| Herpes simplex | Ascending/transplacental | Skin lesions, encephalitis, eye disease | Peripartum most common |
| Varicella | Transplacental | Limb hypoplasia, skin scarring, eye defects, brain atrophy | 1st-2nd trimester |
| Zika virus | Transplacental | Microcephaly, brain malformations | 1st trimester |
| HIV | Transplacental/birth/breast milk | IUGR, opportunistic infections | Any |
CNS propensity: There is a particular propensity for the CNS to be affected by infectious teratogens. The fetal blood-brain barrier (BBB) offers little resistance to microorganisms.
D. Ionizing Radiation
- Exposure to high levels injures embryonic cells → cell death, chromosome breakage, microcephaly, cognitive deficiency, growth restriction
- Severity is dose-, rate-, and stage-dependent
- Period of greatest CNS sensitivity: 8-16 weeks post-fertilization (peak neuronal proliferation)
-
25,000 millirads (mrads) is harmful
- Diagnostic levels (<10,000 mrads): no conclusive proof of harm
- Recommended limit: 500 mrad (0.005 Gy) for entire gestational period
- Diagnostic ultrasound: no confirmed harmful effects
E. Maternal Metabolic and Disease States
| Condition | Teratogenic Effect |
|---|---|
| Diabetes mellitus (poorly controlled) | 2-3× increased birth defects; macrosomia (fat pads over upper back/lower jaw); sacral agenesis, brain anomalies, skeletal defects, CHD; RDS; neonatal hypoglycemia |
| Phenylketonuria (PKU) | Maternal hyperphenylalaninemia → cognitive deficiency, microcephaly, CHD, IUGR in offspring |
| Hypothyroidism | Impaired CNS development (endemic cretinism) |
| Obesity/hyperthermia | Neural tube defects |
F. Chemical/Environmental Teratogens
| Agent | Effect |
|---|---|
| Methylmercury | Cerebral palsy-like picture, deafness, cognitive deficiency (Minamata disease) |
| Polychlorinated biphenyls (PCBs) | IUGR, skin discoloration (main source: contaminated sport fish) |
| Lead | Cognitive deficiency, growth restriction |
PART 3: PRINCIPLES OF TERATOGENESIS
Three fundamental principles govern teratogenicity (Moore & Persaud):
- Critical period of development
- Dose of the agent
- Genotype (genetic constitution) of the embryo
PART 4: MODES OF ACTION OF TERATOGENS
4.1 Cellular Mechanisms
Teratogens act through one or more of these cellular pathways:
| Mechanism | Explanation | Example |
|---|---|---|
| Cell death (apoptosis) | Programmed or pathological cell death during organogenesis | Thalidomide - induces apoptosis in limb bud cells via anti-angiogenic effects and Wnt/IGF signaling disruption |
| Faulty cellular interaction/induction | Disruption of cell signaling between adjacent tissues (paracrine induction) | Retinoic acid - disrupts HOX gene expression domains |
| Reduced biosynthesis of substrates | Impairs synthesis of structural proteins, enzymes, neurotransmitters | Folate antagonists - disrupt nucleotide synthesis → NTDs |
| Impaired morphogenetic movements | Cells fail to migrate correctly (e.g., neural crest cell migration) | Isotretinoin blocks neural crest cell migration |
| Altered gene expression | Disruption of transcription factor cascades (HOX, PAX, NKX genes) | Valproate inhibits histone deacetylase → altered gene expression |
| Mechanical disruption | Physical forces | Amniotic bands, oligohydramnios |
| Impaired DNA repair | Radiation, alkylating agents | Ionizing radiation - DNA double-strand breaks |
| Anti-angiogenesis | Disrupted blood vessel formation in embryo | Thalidomide (anti-VEGF) |
4.2 Molecular Pathways Disrupted
- Retinoic acid (RA) pathway: RA regulates HOX gene expression determining anterior-posterior body patterning. Excess RA from isotretinoin massively upregulates retinoic acid receptors → reprograms HOX boundaries → craniofacial and limb malformations
- Sonic Hedgehog (SHH) pathway: SHH is critical for CNS septation, limb patterning, gut development. Cyclopamine (plant alkaloid) blocks SHH → cyclopia and holoprosencephaly. Defects in cholesterol biosynthesis (e.g., Smith-Lemli-Opitz syndrome) impair SHH signaling because SHH requires cholesterol modification for activation
- WNT pathway: Disruption affects neural crest cell fate and cardiac septation
- Folate-dependent methylation: Folate deficiency → impaired DNA synthesis and methylation → neural tube defects. Supplementation with 400-800 mcg/day prevents up to 70% of NTDs
- Common final pathway: Different teratogens act through different initial mechanisms but converge on a common downstream pathway → intrauterine death, developmental defects, FGR, or functional disturbances
4.3 The "All-or-Nothing" Principle (Weeks 1-2)
During the first 2 weeks post-fertilization (pre-embryonic period), teratogens either:
- Kill the embryo (all-or-nothing effect), OR
- Their disruptive effects are completely compensated by the powerful regulatory properties of the early embryo (totipotency of blastomeres)
Birth defects are NOT produced during weeks 1-2 even if the embryo survives.
PART 5: CRITICAL PERIODS OF HUMAN DEVELOPMENT
This is the most clinically important concept in teratology.
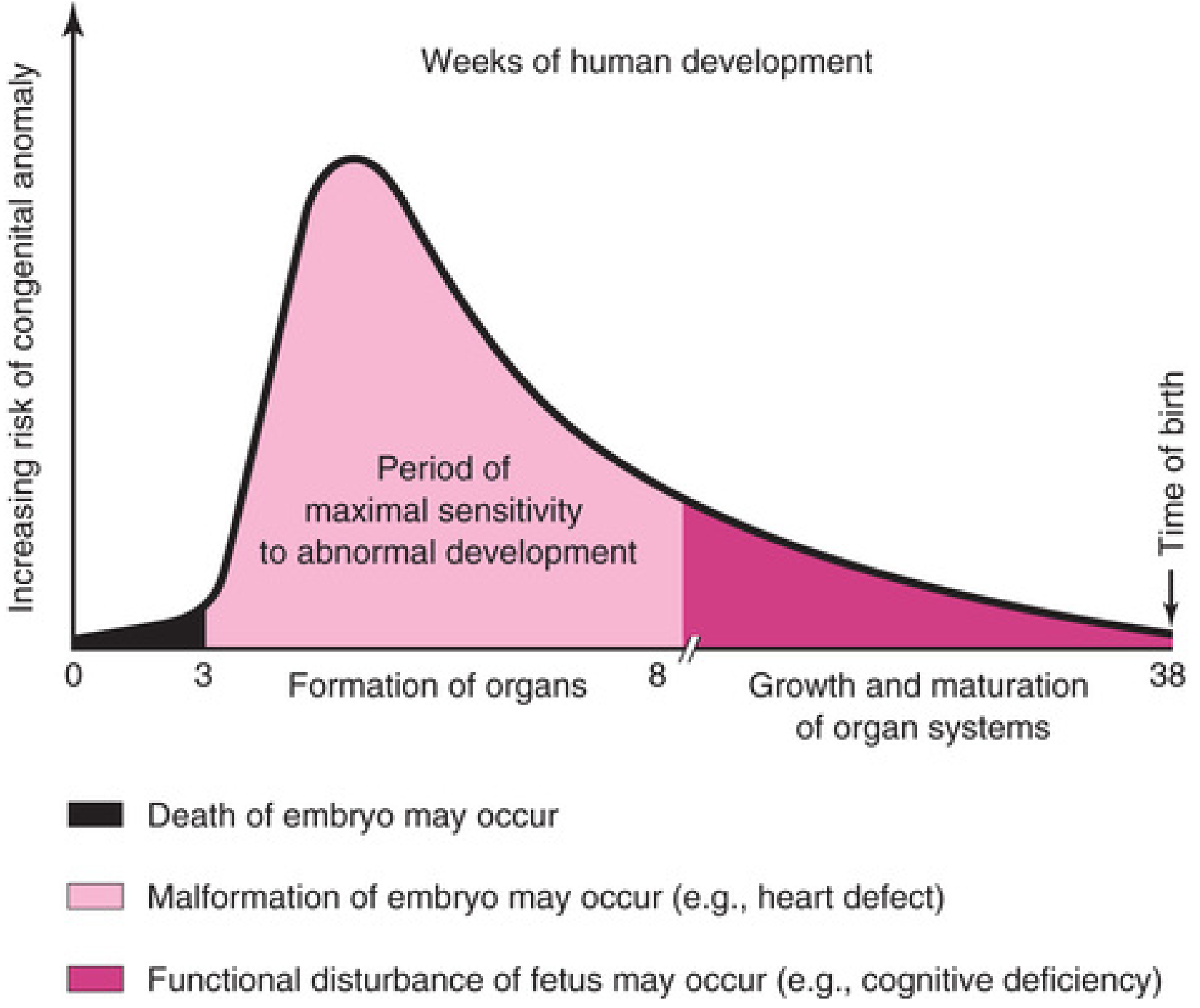
Key principle: The embryo's stage of development when it encounters a teratogen determines susceptibility. The most critical period is when cell division, cell differentiation, and morphogenesis are at their peak.
Critical Period Timeline
| Period | Weeks Post-Fertilization | What Happens | Teratogen Effect |
|---|---|---|---|
| Pre-embryonic | Weeks 1-2 | Cleavage, implantation | Death OR no effect (all-or-nothing) |
| Embryonic (peak sensitivity) | Weeks 3-8 | Organogenesis - all major organ systems form | Major structural malformations |
| Early fetal | Weeks 9-16 | Organ growth and differentiation | Physiologic defects, minor morphologic defects, beginning of functional disruption |
| Mid to late fetal | Weeks 17-38 | Growth and maturation | Functional disturbances (cognitive deficiency, behavioral) - no major structural defects after organogenesis |
Organ-Specific Critical Periods
| Organ/System | Critical Period | Teratogen Timing Matters |
|---|---|---|
| Central nervous system | Weeks 3-16 (brain continues to week 16+, always sensitive) | Any teratogen active 3-16 weeks can cause cognitive deficiency; severe structural defects 3-8 weeks |
| Heart | Weeks 3-8 | Heart defects caused only during this window; heart accounts for 8/1000 birth defects |
| Eyes | Weeks 4-8 | Cataracts, microphthalmia (rubella in week 4-6) |
| Limbs | Weeks 4-8 | Thalidomide days 20-36; limb reduction defects only if teratogen acts before end of limb critical period |
| Ears | Weeks 4-10 | Internal ear: 4-8 weeks; external ear: longer |
| Palate | Weeks 6-10 | Cleft palate from phenytoin, corticosteroids |
| Brain | Weeks 3-16 and beyond | 8-16 weeks is most sensitive for radiation cognitive damage (peak neuronal proliferation) |
| Teeth | Week 14 through 8 years postnatal | Tetracycline can damage both primary and permanent teeth over prolonged window |
| Skeleton | Prolonged, into childhood | |
| Genitalia | Weeks 7-14 | Androgens, DES; male genital tract DES effect before week 11 |
Incidence of major birth defects by organ (at birth):
- Brain: 10/1000
- Heart: 8/1000
- Kidneys: 4/1000
- Limbs: 2/1000
- Other: 6/1000
- Total: ~30/1000 (3%)
Important Corollary
"Because biochemical differentiation precedes morphologic differentiation, the period during which structures are sensitive to interference by teratogens often precedes the stage of their visible development by a few days." - Moore & Persaud
This means teratogenic damage can occur before the organ is even morphologically visible - a critical fact for advising pregnant patients about early drug exposures.
PART 6: TETRALOGY OF FALLOT (ToF)
6.1 Definition and Anatomy
Tetralogy of Fallot is the most common cyanotic congenital heart disease beyond infancy, accounting for approximately 5% of all congenital cardiac malformations.
The four cardinal features:
- Right ventricular outflow tract obstruction (RVOTO) - subpulmonic (infundibular) stenosis is most common; can also be at valvular level or complete atresia
- Large, unrestrictive VSD - typically perimembranous, in the vicinity of the membranous interventricular septum
- Overriding aorta - aortic valve lies immediately over the VSD, receiving blood from both ventricles
- Right ventricular hypertrophy - secondary to high pressure load from RVOTO
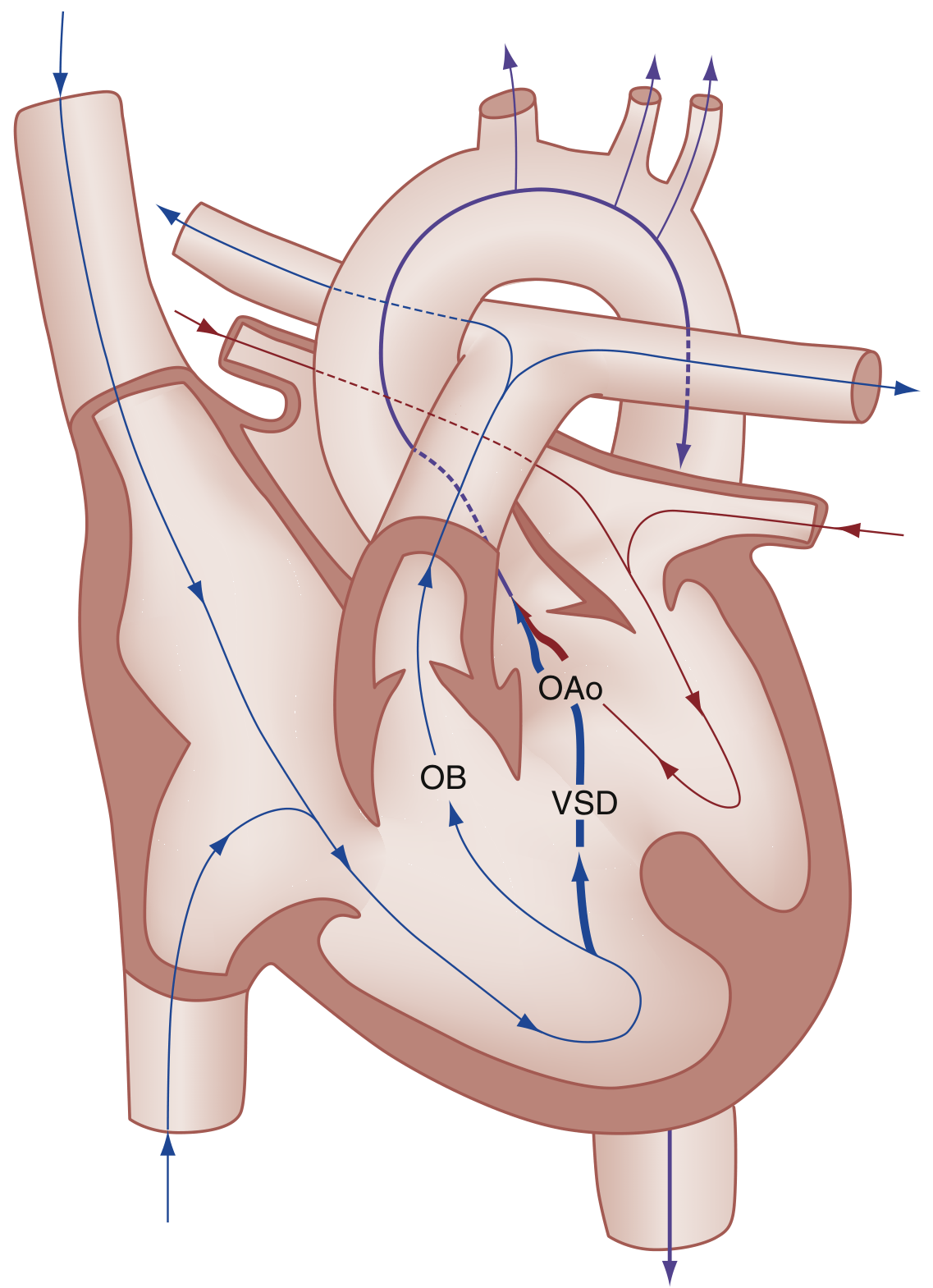
6.2 Embryological Basis
All four features result from a single embryologic defect: anterosuperior displacement of the infundibular (conal/outlet) septum during cardiac development.
- This leads to abnormal septation between the pulmonary trunk and aortic root
- The pulmonary trunk becomes small/hypoplastic
- The aorta overrides the malaligned VSD
- Right ventricular hypertrophy is secondary to RVOTO
Extreme form: Pulmonary atresia with VSD - entire right ventricular output goes through the aorta; pulmonary blood flow depends entirely on PDA or bronchial collateral vessels.
6.3 Gross Pathology
- Heart is enlarged and "boot-shaped" (coeur en sabot) - due to RV hypertrophy
- Proximal aorta: dilated
- Pulmonary trunk: hypoplastic
- RV wall: markedly hypertrophied (can exceed LV thickness)
- VSD: large, near membranous septum
- Left-sided chambers: normal size (important - distinguishes from other cyanotic lesions)
6.4 Hemodynamics and Clinical Presentation
The key clinical determinant is the degree of RVOTO:
| RVOTO Severity | Hemodynamics | Clinical Picture |
|---|---|---|
| Mild ("Pink Tet") | Left-to-right shunt predominates (high left pressure overcomes) | Acyanotic, acts like isolated VSD |
| Moderate | Bidirectional shunting | Mild cyanosis, progressive |
| Severe | Right-to-left shunt predominates | Profound cyanosis within first days of life; PGE1 dependent |
Physical Examination:
- Varying degrees of cyanosis
- Systolic ejection murmur along the left sternal border (from RVOTO, not VSD - the VSD is silent because it is large and non-restrictive)
- With pulmonary atresia: no murmur (no flow through RVOT), continuous murmur from PDA or collaterals
- Chronic hypoxemia → compensatory polycythemia and clubbing of fingers and toes
Investigations:
- CXR: Decreased pulmonary vascular markings ("black lung fields") + boot-shaped heart (concave main pulmonary artery segment on left heart border) + right-sided aortic arch in 25%
- ECG: Right ventricular hypertrophy + right axis deviation
- Echo: Diagnostic - shows RVOTO, VSD, overriding aorta, RVH; measures gradients
6.5 The Tet Spell (Hypercyanotic/Hypoxic Spell)
Peak incidence: 2-4 months of age
Trigger: Any event that suddenly lowers systemic vascular resistance (SVR) - crying, defecation, agitation, feeding, fever, hypovolemia, tachycardia
Vicious cycle:
↓SVR → ↑Right-to-left shunt across VSD → ↓PaO₂, ↑PCO₂, ↓pH → stimulates respiratory center → hyperpnea → ↑negative intrathoracic pressure → ↑venous return to RV → ↑volume shunted right-to-left → worsening hypoxia → deeper hyperpnea → cycle repeats
Clinical features: Hyperpnea, prolonged crying, increasing cyanosis, limpness → possible syncope, seizure, stroke, or death if untreated
Management of Tet Spell (Harriet Lane / Rosen's):
| Step | Intervention | Mechanism |
|---|---|---|
| 1st | Knee-to-chest position | ↑ SVR + ↓ venous return → ↓ right-to-left shunt |
| 2nd | Supplemental O₂ | Limited value alone; provides some pulmonary vasodilation |
| 3rd | Morphine 0.1-0.2 mg/kg IV/IM | Suppresses respiratory center → breaks hyperpnea cycle; reduces SVR and tachycardia |
| Alt | Fentanyl 1 mcg/kg IV/IM (or 1.5-2 mcg/kg intranasal) | Same as morphine |
| Alt | Midazolam 0.2-0.3 mg/kg intranasal | Anxiolysis, reduces crying |
| 4th | NaHCO₃ 1 mEq/kg IV | Corrects metabolic acidosis |
| 5th | Ketamine 1-2 mg/kg IV / 3-5 mg/kg IM | ↑ SVR + sedation |
| 6th | Propranolol 0.1-0.2 mg/kg IV | ↓ HR, relaxes infundibular spasm |
| 7th | Phenylephrine 0.01-0.02 mg/kg IV | Directly ↑ SVR → reverses shunt direction |
6.6 Associations
- Right-sided aortic arch (25%)
- ASD (in some - "Pentalogy of Fallot")
- Anomalous origin of left coronary artery
- Genetic: ~15% have 22q11.2 deletion (DiGeorge/velocardiofacial syndrome) - screen all ToF patients
- Down syndrome (trisomy 21)
6.7 Management
Neonatal/Infant:
- Severe/duct-dependent ToF: PGE1 (prostaglandin E1) infusion to maintain PDA
- Tet spells: managed as above
- Oral propranolol: for preventing tet spells while awaiting surgery
Surgical:
- Complete intracardiac repair (preferred in early infancy, typically 3-6 months): VSD patch closure + relief of RVOTO (infundibular resection ± transannular patch)
- Palliative shunt (if anatomy not suitable for primary repair): Blalock-Taussig-Thomas (BTT) shunt - subclavian artery to pulmonary artery
Long-term issues post-repair:
- Pulmonary regurgitation (after transannular patch) → RV dilation → arrhythmia, RV failure → may need pulmonary valve replacement in 2nd-3rd decade
- Residual RVOT gradient
- Ventricular arrhythmias (risk of sudden cardiac death - follow with Holter monitoring)
SUMMARY - THE BIG PICTURE
Teratology = Study of abnormal development
TERATOGEN acts during CRITICAL PERIOD
↓
Mode of action:
- Cell death
- Disrupted induction
- Altered gene expression (HOX, SHH, WNT)
- Impaired cell migration
- Mechanical disruption
↓
Outcome depends on:
1. Timing (critical period of organ)
2. Dose
3. Fetal genotype
↓
WEEK 1-2: all-or-nothing (death or no defect)
WEEK 3-8: MAJOR MALFORMATIONS (peak risk)
WEEK 9-16: minor morphologic + beginning functional deficits
WEEK 17-38: FUNCTIONAL deficits only (cognitive, behavioral)
Tetralogy of Fallot = single embryologic defect (infundibular septal malalignment during cardiac organogenesis, weeks 3-7) → 4 anatomic consequences → right-to-left shunt → cyanosis → tet spells → repair in infancy → excellent long-term survival with follow-up.
- The Developing Human: Clinically Oriented Embryology, Chapter 20
- Robbins & Kumar Basic Pathology, Chapter 9
- Rosen's Emergency Medicine, Chapter 165
- The Harriet Lane Handbook, Chapter 13
This is a shared conversation. Sign in to Orris to start your own chat.