Rbc membrane disorders, fragmentation disorders and enzyme deficiency
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
Reading File
I now have comprehensive material from multiple authoritative sources. Let me compile the full detailed answer.
RBC Membrane Disorders, Fragmentation Disorders, and Enzyme Deficiencies
OVERVIEW: Classification of Hemolytic Anemias
Hemolytic anemias are characterized by a shortened RBC lifespan (normal 120 days), with resulting reticulocytosis and erythroid hyperplasia. They are broadly classified as:
- Intracorpuscular (intrinsic) defects: membrane disorders, enzyme deficiencies, hemoglobinopathies
- Extracorpuscular (extrinsic) defects: immune, fragmentation, infections, drugs
Hemolysis may be extravascular (spleen-mediated) or intravascular (within blood vessels). Key labs in all hemolytic anemias include: low haptoglobin, elevated LDH (LD-1 isoform from RBCs), elevated unconjugated bilirubin, and reticulocytosis.
PART 1: RBC MEMBRANE DISORDERS
1A. Hereditary Spherocytosis (HS)
Genetics: Autosomal dominant in ~75% of cases; prevalence ~1 in 5000 in northern Europe.
Normal RBC membrane structure:
The membrane skeleton consists of spectrin (alpha and beta chains forming heterodimers that self-associate into tetramers), actin oligomers, and two anchoring systems:
- Spectrin → ankyrin → band 4.2 → Band 3 (transmembrane ion transporter)
- Spectrin "tail" → Protein 4.1 → Glycophorin A
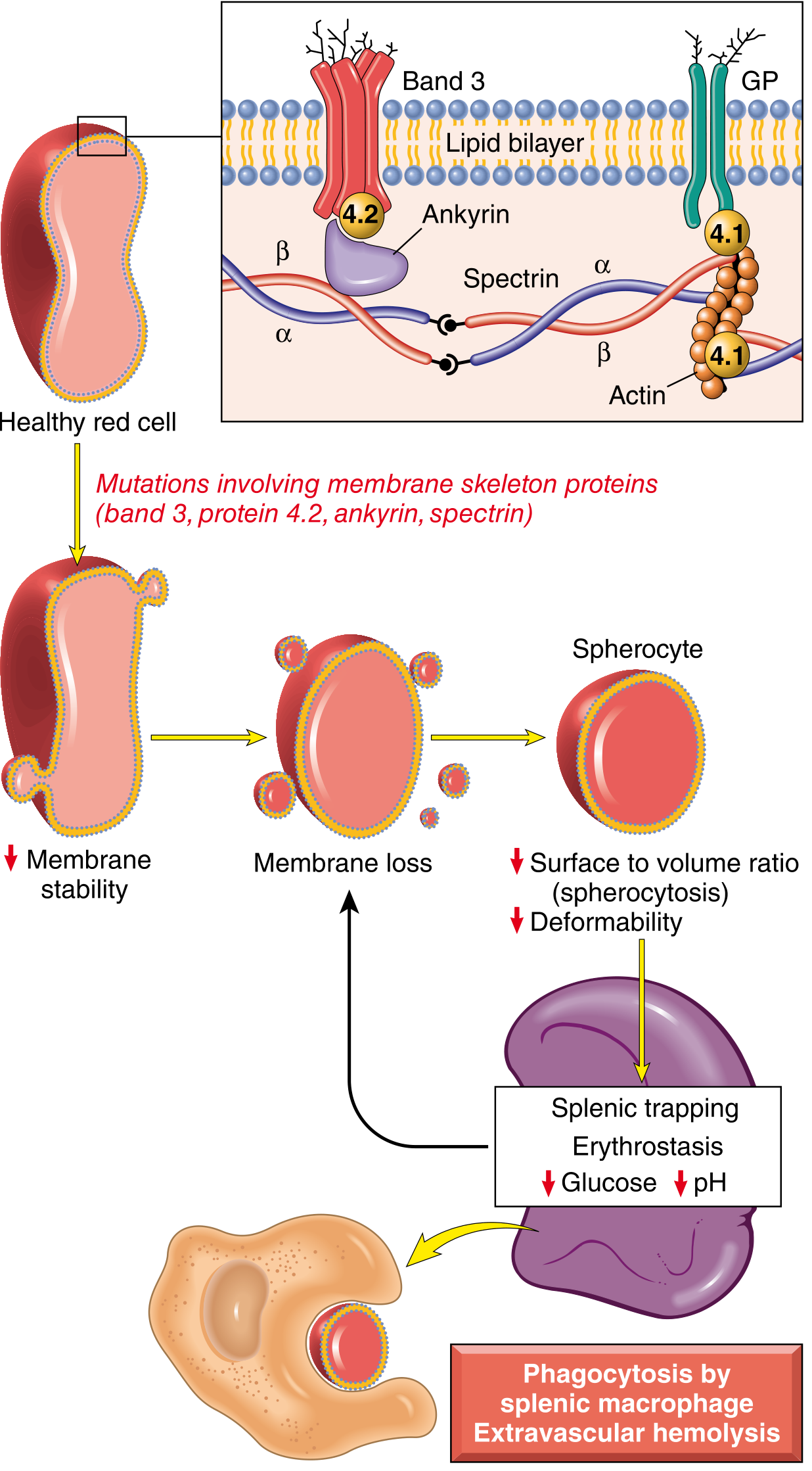
Fig. 14.2 - Pathogenesis of hereditary spherocytosis. Mutations in spectrin, ankyrin, band 3, or band 4.2 destabilize the membrane skeleton, leading to progressive membrane loss, spherocyte formation, and extravascular hemolysis. (Robbins, Cotran & Kumar Pathologic Basis of Disease)
Pathogenesis:
- Mutations (most commonly frameshift/premature stop codons) affect ankyrin, band 3, spectrin (α or β), or band 4.2
- These reduce membrane skeleton assembly, destabilizing the lipid bilayer
- The membrane progressively sheds fragments as RBCs age → decreased surface-area-to-volume ratio → cells adopt the minimal-volume shape: a sphere
- Spherocytes are non-deformable → trapped in splenic red pulp cords → phagocytosed by macrophages (extravascular hemolysis)
- Splenic environment worsens the process: glucose depletion, low pH, and erythrostasis further damage spherocytes
- After splenectomy: spherocytes persist but anemia corrects
Morphology:
- Peripheral smear: small, dark (hyperchromic), uniformly staining spherocytes without central pallor, anisocytosis
- Moderate splenomegaly (500-1000 g)
- Pigment (bilirubin) gallstones in 40-50% of adults
- Bone marrow erythroid hyperplasia
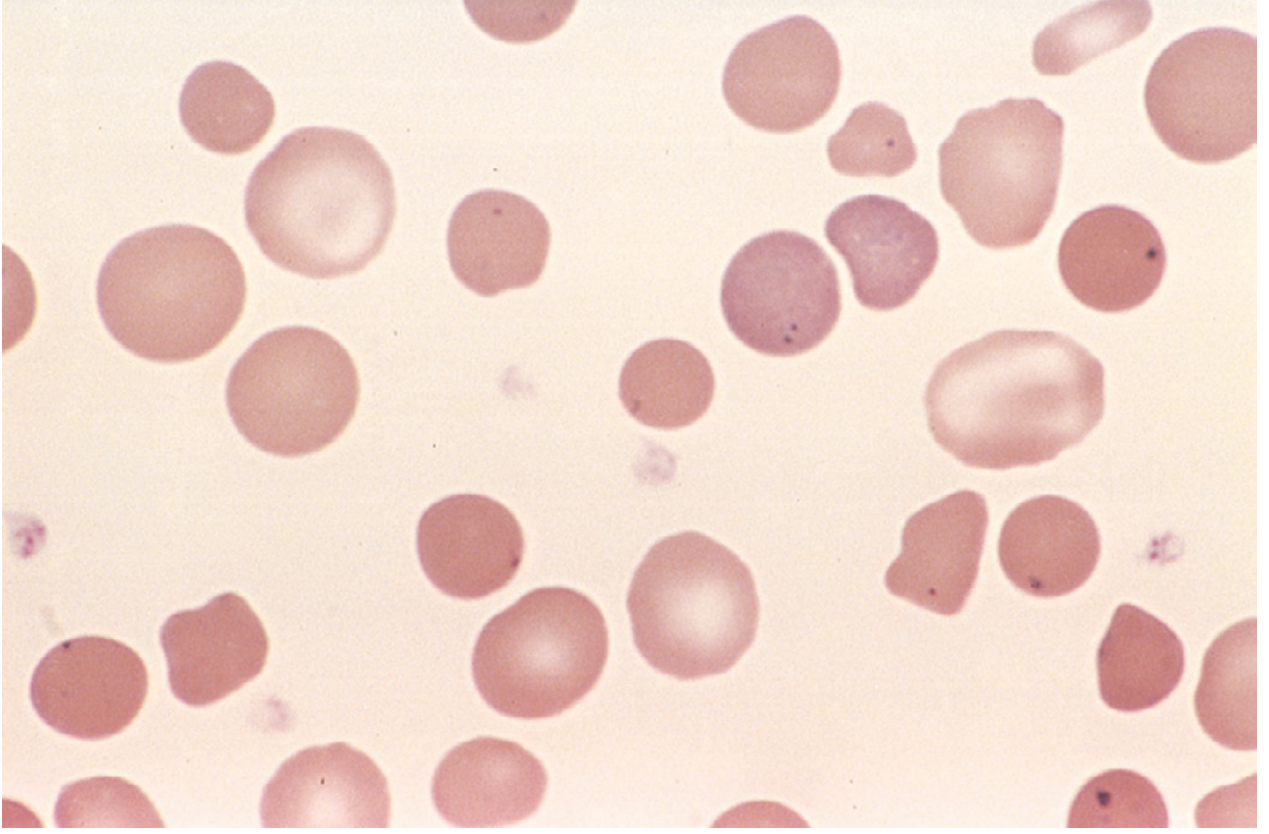
Hereditary spherocytosis - peripheral blood smear. Small dark spherocytes lack central pallor. Howell-Jolly bodies (dark nuclear remnants) visible in this asplenic patient. (Robbins, Cotran & Kumar)
Clinical features:
- Triad: anemia + splenomegaly + jaundice
- Increased MCHC (due to K⁺/H₂O loss = cellular dehydration)
- Aplastic crises triggered by parvovirus B19 (infects erythroid precursors)
- Hemolytic crises with infections
Diagnosis:
- Spherocytes + reticulocytosis + negative direct Coombs test (distinguishes from autoimmune hemolytic anemia)
- Elevated MCHC
- Eosin-5'-maleimide (EMA) binding test: decreased fluorescence (band 3 loss)
- Osmotic fragility test: spherocytes lyse at higher NaCl concentrations than normal cells
Treatment: Splenectomy (corrects anemia, spherocytes persist); folic acid supplementation.
1B. Hereditary Elliptocytosis (HE)
Genetics: Autosomal dominant; prevalence ~1 in 2000-4000. Common in African and Mediterranean ancestry (HE may confer partial malaria resistance).
Pathogenesis:
- Horizontal defect - weakness of the membrane skeleton horizontal lattice
- Principal defects in α-spectrin or β-spectrin → impaired self-association of spectrin dimers into tetramers and oligomers
- Other defects: protein 4.1 (disrupts spectrin-actin junctional complex), glycophorin C
- Elliptical shape reflects failure to maintain normal RBC geometry after shear deformation
Clinical spectrum (heterogeneous):
| Form | Features |
|---|---|
| Common HE (~90%) | Most asymptomatic; elliptocytes >15% on smear; only 10-20% have mild hemolysis |
| Spherocytic HE (~10%) | Mild-moderate hemolytic anemia + splenomegaly; both elliptocytes and spherocytes |
| Southeast Asian ovalocytosis | Often asymptomatic; very rigid oval cells; resistance to cerebral malaria |
| Hereditary Pyropoikilocytosis (HPP) | Severe; micropoikilocytosis + fragmentation; autosomal recessive (homozygous/compound heterozygous spectrin mutations); RBCs fragment at 45-46°C (normal: 49°C) |
Diagnosis: Peripheral smear shows >15% elliptocytes (cigar/rod-shaped); osmotic fragility often normal in common HE.
1C. Paroxysmal Nocturnal Hemoglobinuria (PNH)
Though technically an acquired clonal disorder (not inherited), PNH is a membrane disorder:
Pathogenesis:
- Acquired somatic mutation in PIGA gene (X-linked) in a hematopoietic stem cell
- PIGA is required to synthesize GPI anchors (phosphatidylinositol glycan)
- Loss of GPI-anchored complement-regulatory proteins: CD55 (DAF) and CD59 (MIRL)
- RBCs derived from the mutant clone are exquisitely sensitive to lysis by the complement C5b-C9 membrane attack complex
- Nocturnal hemolysis: complement activation enhanced by CO₂ retention during sleep → mild acidosis → enhanced complement fixation
- Most patients actually present with chronic iron-deficiency anemia from persistent intravascular hemolysis
Complications:
- Thrombosis (most feared): particularly portal vein, hepatic vein (Budd-Chiari), cerebral veins
- Iron deficiency from hemoglobinuria
- Association with aplastic anemia
Treatment:
- Eculizumab (anti-C5 monoclonal antibody): blocks MAC formation, reduces intravascular hemolysis and thrombosis
- Caveat: eculizumab increases risk of Neisseria infections (meningococcal vaccination mandatory)
- Ravulizumab (long-acting anti-C5); iptacopan (factor B inhibitor, targets upstream complement)
Diagnosis: Flow cytometry showing absence of CD55/CD59 on RBCs and granulocytes; Ham's test (historical).
PART 2: FRAGMENTATION (MICROANGIOPATHIC) HEMOLYTIC ANEMIAS
Fragmentation anemias involve the mechanical destruction of RBCs, producing schistocytes (helmet cells, triangular fragments) on the peripheral smear. The mechanism is physical shearing of RBCs by fibrin strands, platelet thrombi, or turbulent flow.
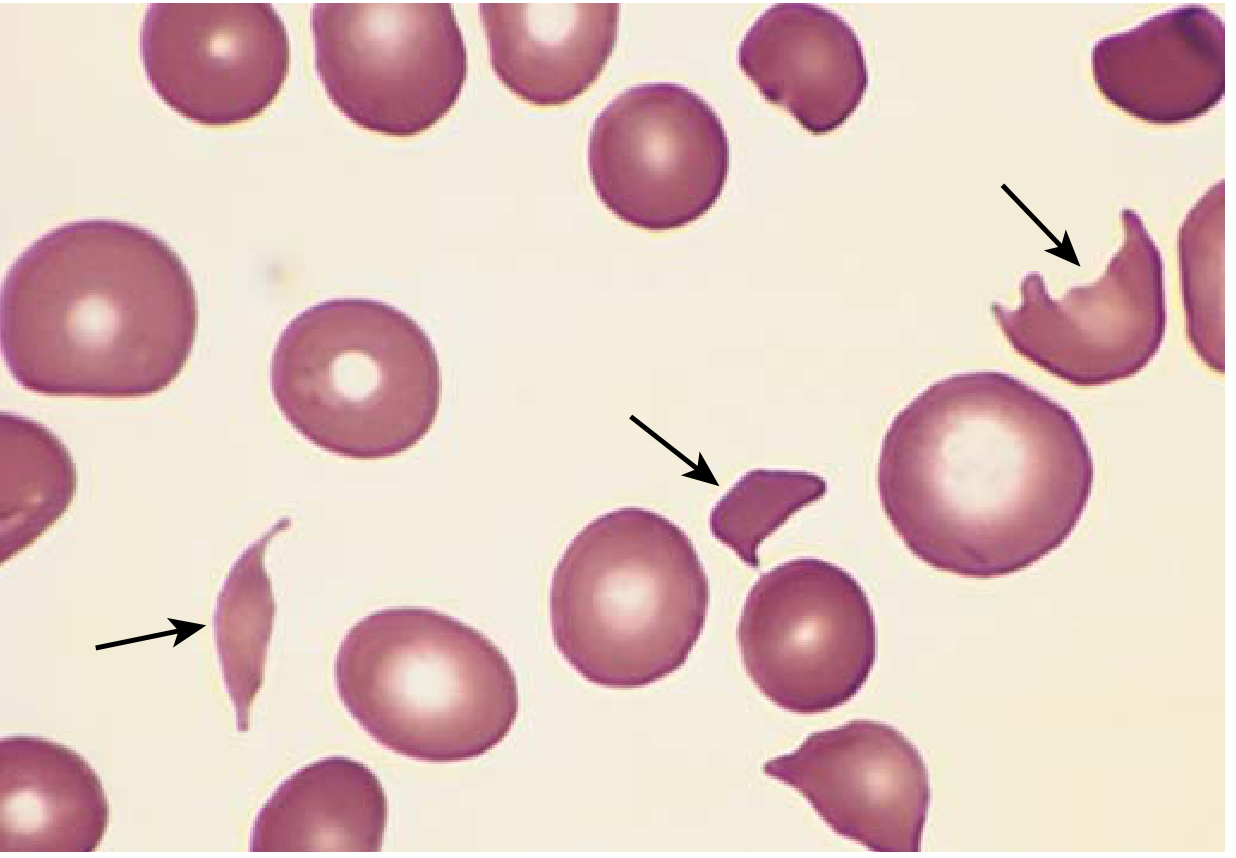
Microangiopathic hemolytic anemia - peripheral smear. Multiple fragmented red cells (schistocytes, arrows) in a patient with HUS. (Robbins & Kumar Basic Pathology)
Causes of Fragmentation Hemolysis
| Category | Examples | Mechanism |
|---|---|---|
| TTP (Thrombotic Thrombocytopenic Purpura) | ADAMTS13 deficiency/inhibition | Platelet thrombi in microvasculature shear RBCs |
| HUS (Hemolytic Uremic Syndrome) | E. coli O157:H7, S. pneumoniae, Shigella | Bacterial toxin induces platelet aggregation + endothelial injury |
| DIC | Sepsis, malignancy, obstetric catastrophes | Fibrin deposition in microvasculature sheaves RBCs |
| Malignant hypertension | - | Fibrin deposition in arterioles |
| Prosthetic heart valve | Mechanical valves, paravalvular leaks | Turbulent flow fractures RBCs |
| Bone marrow microangiopathy | Metastatic cancer, leukemia, lymphoma, myelofibrosis | Space-occupying lesions → RBC destruction in marrow microvasculature |
| Immune-mediated | SLE, anti-phospholipid syndrome | Endothelial damage → immune complex + fibrin deposition |
| March hemoglobinuria | Repetitive foot-strike | Physical trauma to RBCs in plantar capillaries |
| Burns | Severe thermal injury | Direct heat denaturation of RBC membrane |
TTP - Key Details
- Caused by deficiency or inhibition (autoantibody) of ADAMTS13 - a metalloprotease that cleaves ultra-large von Willebrand factor (vWF) multimers
- Uncleaved high-molecular-weight vWF multimers promote platelet aggregation → platelet thrombi in microvasculature
- Pentad: Microangiopathic hemolytic anemia + thrombocytopenia + fever + renal failure + neurological symptoms
- Treatment: Plasma exchange (replaces ADAMTS13 and removes inhibitory antibodies)
- Caplacizumab (anti-vWF nanobody): adjunct therapy
HUS - Key Details
- Mainly children; often follows hemorrhagic diarrhea with enterohemorrhagic E. coli O157:H7 (Shiga toxin)
- Shiga toxin → endothelial injury, especially in glomeruli → microangiopathic hemolysis + thrombocytopenia + acute renal failure (predominant)
- Triad: Microangiopathic hemolytic anemia + thrombocytopenia + acute kidney injury
- Atypical HUS: complement dysregulation (mutations in complement factor H, I, or CD46)
Leukoerythroblastic Picture
When fragmentation occurs within the bone marrow microvasculature (myelophthisic process), both RBC and WBC precursors are released into circulation:
- Nucleated RBCs + myelocytes/metamyelocytes = leukoerythroblastic picture
- Suggests space-occupying bone marrow lesion (metastases, myelofibrosis)
PART 3: RBC ENZYME DEFICIENCIES
RBCs are metabolically anucleate - they cannot synthesize new enzymes. Two key metabolic pathways protect RBCs:
- Embden-Meyerhof (glycolytic) pathway → produces ATP (for membrane pumps, deformability)
- Hexose monophosphate (HMP) shunt / Pentose phosphate pathway → produces NADPH → reduced glutathione (GSH) → oxidant defense
3A. G6PD Deficiency (Most Common RBC Enzyme Defect)
Gene: X-linked (Xq28), therefore predominantly affects males
Prevalence: Most common RBC enzyme defect worldwide; highest in Africa, Mediterranean, Middle East, South/Southeast Asia
Pathogenesis:
- G6PD catalyzes the first step of the HMP shunt: Glucose-6-phosphate → 6-phosphogluconate + NADPH
- NADPH is the sole electron donor for RBC oxidant defense (reduces GSH via glutathione reductase)
- In G6PD deficiency: oxidant stress → GSH cannot be regenerated → hemoglobin oxidizes → denaturation → Heinz bodies (intracellular precipitates) form
- Heinz bodies damage the membrane → intravascular hemolysis
- Splenic macrophages attempt to "pluck out" Heinz bodies → bite cells (keratocytes)
- Cells with reduced deformability are trapped and destroyed in the spleen
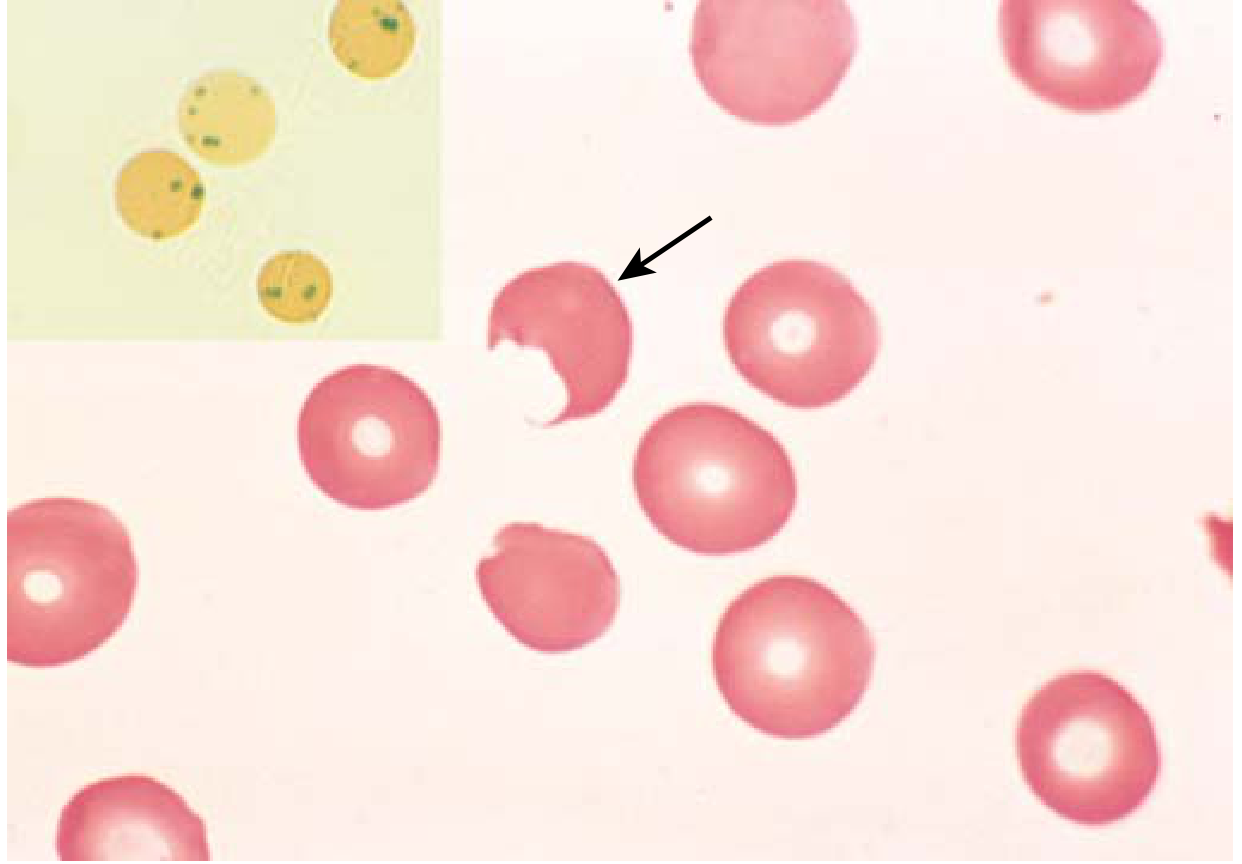
G6PD deficiency - peripheral smear. The "bite cell" (arrow) is created when splenic macrophages pluck out Heinz bodies. Inset: Heinz bodies on supravital stain. (Robbins & Kumar Basic Pathology)
Triggers of hemolysis:
- Drugs: Primaquine, dapsone, sulfonamides, nitrofurantoin, rasburicase, phenacetin, high-dose aspirin, vitamin K analogues
- Foods: Fava beans (favism) - contain divicine and isouramil
- Infection: Most common trigger; phagocytes generate H₂O₂ as host response → oxidant load
- Metabolic: Diabetic ketoacidosis
Clinically important variants:
| Variant | Population | Severity | Notes |
|---|---|---|---|
| G6PD A- | Sub-Saharan Africa | Mild | Enzyme half-life modestly reduced; only old RBCs affected; hemolysis self-limiting (young RBCs with adequate G6PD replace them) |
| G6PD Mediterranean | Mediterranean, Middle East | Severe | More profound enzyme deficiency; older AND younger RBCs affected; hemolysis more severe and prolonged |
| G6PD Canton | Southeast Asia | Moderate-severe | - |
| G6PD B (normal) | Universal wild-type | - | Baseline |
X-linkage and gender:
- Affected males: ALL RBCs are G6PD deficient (uniform deficiency)
- Carrier females (heterozygotes): X-inactivation (lyonization) creates two populations of RBCs - normal and G6PD-deficient. Most are unaffected, but "unfavorable lyonization" (majority of cells inactivate the normal X) can produce disease. G6PD deficiency is not truly recessive in females due to X-inactivation mosaicism.
Clinical course:
- Episodic intravascular hemolysis 2-3 days after trigger
- Hemoglobinuria (dark urine), jaundice, anemia
- G6PD A- variant: hemolysis is self-limiting (young RBCs replenish G6PD)
- Diagnosis: Fluorescent spot test (NADPH fluoresces under UV); enzyme assay (must test when patient NOT in crisis - new reticulocytes have normal G6PD)
- Neonatal jaundice is common in G6PD-deficient newborns
3B. Pyruvate Kinase (PK) Deficiency
Gene: PKLR gene; chromosome 1q22; autosomal recessive
Prevalence: ~1:20,000 in the general white population; second most common RBC enzyme defect (after G6PD)
Pathogenesis:
- PK catalyzes a key ATP-generating step in glycolysis: phosphoenolpyruvate (PEP) + ADP → pyruvate + ATP
- ATP deficiency → failure of Na⁺/K⁺-ATPase → cellular dehydration → rigid, poorly deformable RBCs → extravascular hemolysis (spleen-mediated)
- Glycolytic block → accumulation of 2,3-DPG (2,3-bisphosphoglycerate) upstream of the PK step
- High 2,3-DPG shifts the O₂-Hgb dissociation curve rightward → better O₂ delivery to tissues
- This is why patients tolerate the anemia relatively well (tissue hypoxia is less than the Hgb level would suggest)
- No Heinz bodies (NOT an oxidative hemolysis)
-
300 mutations in PKLR; most affected patients are compound heterozygotes
Features:
- Congenital nonspherocytic hemolytic anemia (chronic, Coombs-negative)
- Hemoglobin typically 6.5-11 g/dL
- Splenomegaly in 80-85%; pigment gallstones in 30-45%
- Neonatal jaundice in >50% (may require exchange transfusion)
- Peripheral smear: often unremarkable; after splenectomy → echinocytes, crenated/irregularly contracted cells
- Reticulocyte count elevated; paradoxically increases further after splenectomy (reticulocytes trapped in spleen are released)
Diagnosis:
- Fluorescent spot test: pyruvate reduces NADH → NAD (loss of fluorescence); failure of fluorescence loss = PK deficient
- Quantitative PK assay at low PEP concentrations (mutant PK may have normal activity at high PEP)
- Autohemolysis test: type II pattern (not prevented by glucose)
- Must remove WBCs before assay (WBCs contain 300× more PK than RBCs)
Treatment:
- Folic acid supplementation (chronic hemolysis → increased folate demand)
- Splenectomy: modest increase in Hgb (~1-2 g/dL); reticulocytes increase sharply
- Iron chelation (iron overload even without transfusion, from increased intestinal absorption driven by erythropoietic drive)
- Mitapivat: Allosteric PK activator; first approved drug for a RBC enzymopathy; effective in patients with missense mutations (raises Hgb significantly in ~50% of patients)
- Bone marrow transplantation for severe cases; gene therapy under investigation
3C. Other Glycolytic Enzyme Deficiencies
All are rare to very rare, autosomal recessive, and cause chronic Coombs-negative hemolytic anemia:
| Enzyme | Pathway | Key Extra-RBC Features |
|---|---|---|
| Hexokinase | Glycolysis | - |
| Glucose-6-phosphate isomerase | Glycolysis | Neuromuscular involvement |
| Phosphofructokinase (PFK) | Glycolysis | Myopathy, myoglobinuria (Tarui disease) |
| Aldolase | Glycolysis | Myopathy |
| Triose phosphate isomerase (TPI) | Glycolysis | Severe CNS involvement (intellectual disability), neuromuscular disease |
| Phosphoglycerate kinase (PGK) | Glycolysis | X-linked; CNS + neuromuscular |
| Glutathione synthase | HMP/Redox | CNS |
| Glutathione reductase | HMP/Redox | Cataracts; hemolysis from flavism |
General approach to glycolytic enzymopathies:
- Unlike membrane disorders, morphologic abnormalities are usually absent
- Coombs-negative HA → suspect enzymopathy
- Definitive diagnosis requires quantitative enzyme assays (specialized labs)
- Management principles similar to PK deficiency: folic acid, transfusions, splenectomy, iron chelation as needed
Summary Comparison Table
| Feature | HS | HE | PNH | Fragmentation (MAHA) | G6PD Deficiency | PK Deficiency |
|---|---|---|---|---|---|---|
| Type | Membrane | Membrane | Membrane (acquired) | Extrinsic/Mechanical | Enzyme (HMP shunt) | Enzyme (Glycolysis) |
| Inheritance | AD (75%) | AD | Acquired (PIGA mutation) | - | X-linked | AR |
| Hemolysis | Extravascular | Extravascular | Intravascular | Intravascular | Intravascular ± extravascular | Extravascular |
| Smear | Spherocytes (no central pallor) | Elliptocytes | Normal or macrocytes | Schistocytes | Bite cells, Heinz bodies | Echinocytes (post-splenectomy) |
| Key defect | Ankyrin/Spectrin/Band 3/Band 4.2 | Spectrin dimer self-association | PIGA → loss of CD55/CD59 | Fibrin/platelet thrombi shear RBCs | NADPH/GSH deficiency | ATP deficiency |
| Trigger | None (chronic) | None (chronic) | Complement activation | Underlying disease (TTP/HUS/DIC/valve) | Drugs/infection/fava beans | None (chronic) |
| Coombs test | Negative | Negative | Negative | Negative | Negative | Negative |
| Splenomegaly | Yes | Sometimes | No | No | No | Yes |
| Thrombocytopenia | No | No | Possible | Yes (TTP, HUS, DIC) | No | No |
| Specific test | EMA binding, osmotic fragility | Smear | Flow cytometry (CD55/CD59) | PT/aPTT (if DIC), ADAMTS13 | Fluorescent spot test; enzyme assay | Fluorescent spot test; PK assay |
| Treatment | Splenectomy | Splenectomy (if severe) | Eculizumab | Treat underlying cause; plasma exchange (TTP) | Avoid triggers | Mitapivat; splenectomy |
Sources:
- Robbins, Cotran & Kumar - Pathologic Basis of Disease (9780443264528)
- Robbins & Kumar Basic Pathology (9780323790185)
- Harrison's Principles of Internal Medicine 22E (9781265977061)
- Henry's Clinical Diagnosis and Management by Laboratory Methods (9780323673204)
- Goldman-Cecil Medicine (9780323930345)
This is a shared conversation. Sign in to Orris to start your own chat.