Inflammation in detail
Reading File
Finding Sources
Finding Sources
Searching PubMed
"inflammation"[MeSH Terms] AND "review"[pt]
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Searching Images
acute inflammation vascular changes leukocyte migration
Reading File
Reading File
Reading File
Reading File
Reading File
I now have rich, comprehensive content from Robbins to construct a thorough, detailed answer on inflammation. Let me compile this now.
Inflammation — A Detailed Overview
Based on: Robbins & Kumar Basic Pathology (10th ed.) and Robbins, Cotran & Kumar Pathologic Basis of Disease
1. Definition and Purpose
Inflammation is a protective vascular and cellular response of living, vascularized tissue to injury, infection, or abnormal stimuli. Its goal is to eliminate the cause of cell injury (microbes, toxins, necrotic cells), remove damaged tissue, and initiate repair. Despite being fundamentally protective, inflammation can itself cause disease when poorly regulated (e.g., rheumatoid arthritis, atherosclerosis, asthma).
The cardinal signs of inflammation, described since antiquity:
| Latin | English |
|---|---|
| Rubor | Redness |
| Tumor | Swelling |
| Calor | Warmth |
| Dolor | Pain |
| Functio laesa | Loss of function (added by Virchow) |
2. Classification: Acute vs. Chronic
| Feature | Acute | Chronic |
|---|---|---|
| Onset | Minutes to hours | Weeks to months |
| Duration | Short lived | Prolonged |
| Dominant cell | Neutrophil | Macrophage, lymphocyte, plasma cell |
| Exudate | Fluid/protein rich | Little exudate |
| Tissue damage | Limited | Tissue destruction + fibrosis |
| Outcome | Resolution, scar, or → chronic | Fibrosis, amyloidosis |
3. Acute Inflammation
3.1 Three Cardinal Components
- Vasodilation of small vessels → redness and warmth
- Increased vascular permeability → exudate formation
- Leukocyte emigration from the microcirculation to the site of injury
All changes occur predominantly in postcapillary venules.
3.2 Vascular Changes
Vasodilation is the first reaction, mediated primarily by histamine (released from mast cells). It causes increased blood flow, producing erythema and warmth.
Increased vascular permeability follows rapidly. Vascular leakage is caused by:
- Endothelial cell contraction creating interendothelial gaps — triggered by histamine, bradykinin, leukotrienes; occurs within 15–30 minutes and is short lived
- Direct endothelial injury (e.g., burns) — immediate and sustained leakage
The result is exudation — escape of protein-rich fluid into the interstitium:
An exudate is a protein-rich extravascular fluid containing cellular debris, indicating increased vascular permeability. A transudate is a protein-poor ultrafiltrate formed without increased permeability (osmotic/hydrostatic imbalance). Pus (purulent exudate) is rich in neutrophils and dead cell debris.
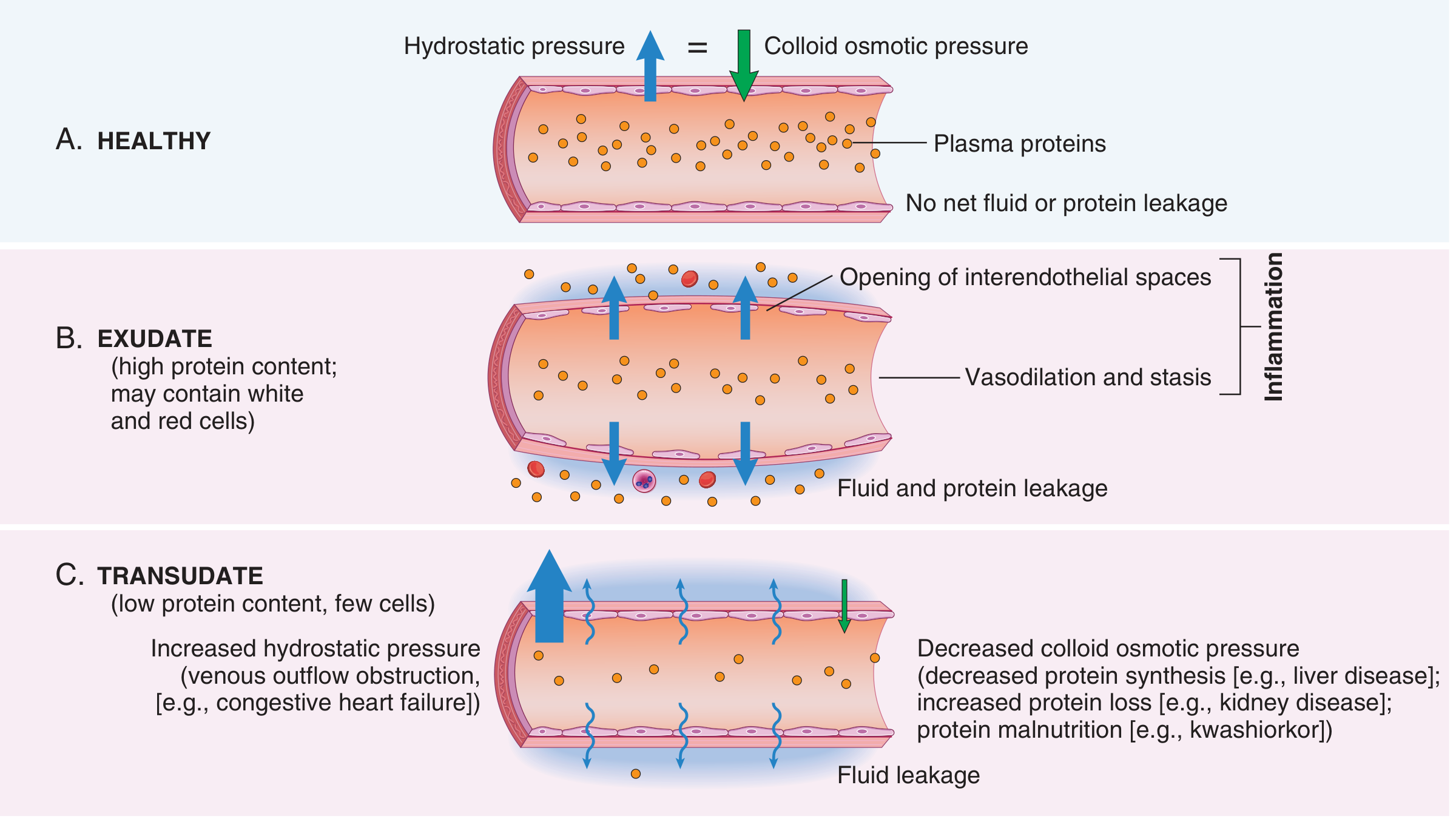
Stasis develops as fluid leaves vessels, blood viscosity rises (hemoconcentration), and flow slows → stage is set for leukocyte margination.
3.3 Leukocyte Recruitment — The Multistep Process
Leukocyte emigration to the site of injury is a highly orchestrated, stepwise process:
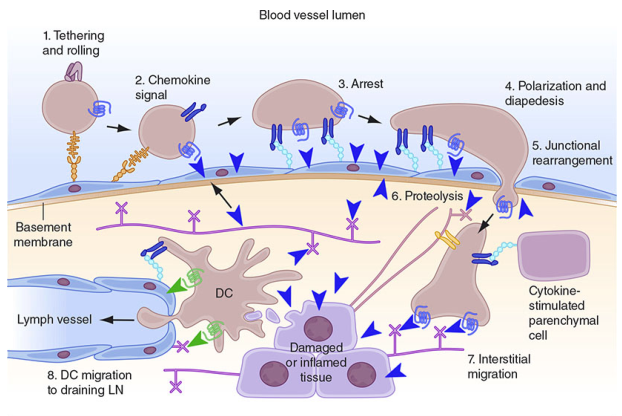
Steps:
- Margination — Leukocytes move to vessel periphery as flow slows
- Rolling — Loose adhesion via selectins (P- and E-selectin on activated endothelium; L-selectin on leukocytes); leukocytes tumble along the endothelial surface
- Firm adhesion — Triggered by chemokines activating leukocyte integrins (LFA-1, Mac-1) which bind ICAM-1 on endothelium
- Transmigration (diapedesis) — Leukocytes squeeze through inter-endothelial junctions mediated by PECAM-1 (CD31)
- Migration in tissues — Directed movement toward the site of injury along a chemical gradient (chemotaxis) — key chemoattractants: C5a, LTB4, IL-8 (CXCL8), bacterial peptides (f-Met-Leu-Phe)
| Step | Key Molecules |
|---|---|
| Rolling | Selectins (P-, E-, L-) |
| Activation | Chemokines (IL-8, C5a) |
| Firm adhesion | Integrins (LFA-1/Mac-1) ↔ ICAM-1 |
| Transmigration | PECAM-1 (CD31) |
3.4 Leukocyte Functions at the Site of Injury
Phagocytosis
- Recognition and attachment — Enhanced by opsonins (IgG, C3b)
- Engulfment — Pseudopod formation and phagosome creation
- Killing and degradation — Via reactive oxygen species (ROS) and lysosomal enzymes
Intracellular Killing Mechanisms
- Oxygen-dependent (respiratory burst): NADPH oxidase generates superoxide (O₂⁻) → hydrogen peroxide (H₂O₂) → hypochlorous acid (HOCl) via myeloperoxidase (MPO) — most potent bactericidal mechanism
- Oxygen-independent: Lysozyme, lactoferrin, defensins, elastase, cathepsins (from granules)
Neutrophil Extracellular Traps (NETs)
Activated neutrophils can release chromatin + histones + antimicrobial peptides as fibrillar extracellular networks that trap and kill microbes extracellularly. NET formation kills the neutrophil in the process and is also detected in sepsis.
Leukocyte-Mediated Tissue Injury
Leukocytes can damage normal host tissues when:
- Defending against resistant organisms (e.g., mycobacteria)
- Directed against self antigens (autoimmune) or harmless antigens (allergy)
- Encountering indigestible material (e.g., urate crystals, silica)
Proteases are controlled by antiproteases (notably α₁-antitrypsin); deficiency → sustained tissue destruction.
3.5 Outcomes of Acute Inflammation
- Complete resolution — Debris cleared by macrophages; edema reabsorbed via lymphatics; tissue returns to normal (when injury is limited and parenchymal cells can regenerate)
- Healing by connective tissue replacement (fibrosis/scar) — When tissue is incapable of regeneration, or abundant fibrin exudate cannot be cleared
- Progression to chronic inflammation — When injurious agent persists or normal healing is disrupted
4. Mediators of Inflammation
Inflammatory mediators are produced at the site of injury and regulate all components of the inflammatory response.
General principles:
- Derived from plasma (synthesized in liver as inactive precursors, activated at the site) or from cells (mast cells, platelets, macrophages, neutrophils, endothelium)
- Most have short half-lives — they are degraded, inactivated, or become exhausted
- Triggering one mediator often activates others (cascade/amplification)
Key Mediators
| Mediator | Source | Main Actions |
|---|---|---|
| Histamine | Mast cells, basophils, platelets | Vasodilation, ↑ vascular permeability (early, rapid) |
| Serotonin (5-HT) | Platelets | Vasodilation, ↑ permeability |
| Prostaglandins (PGE₂, PGI₂) | Arachidonic acid via COX | Vasodilation, pain sensitization, fever |
| Leukotrienes (LTB₄) | Arachidonic acid via LOX | LTB₄: chemotaxis; LTC₄, LTD₄, LTE₄: ↑ permeability, bronchoconstriction |
| PAF (Platelet-activating factor) | Leukocytes, endothelium | ↑ Permeability, leukocyte activation, bronchoconstriction |
| C3a, C5a | Complement activation | ↑ Vascular permeability (C3a), opsonization (C3b), chemotaxis (C5a) |
| Bradykinin | Kinin system (Hageman factor XII) | ↑ Permeability, pain, vasodilation |
| Thrombin | Coagulation cascade | ↑ Permeability, leukocyte and endothelial activation |
| TNF-α, IL-1 | Macrophages, endothelium | Fever, leukocyte activation, endothelial upregulation of adhesion molecules; systemic acute-phase response |
| IL-6 | Macrophages, T cells | Acute-phase protein synthesis (liver) |
| IL-8 (CXCL8) | Macrophages, endothelium | Potent neutrophil chemotaxis |
| IFN-γ | T lymphocytes | Macrophage activation (M1 polarization) |
| NO (nitric oxide) | Endothelium, macrophages | Vasodilation; microbicidal |
| ROS | Neutrophils, macrophages | Microbicidal; also tissue damage |
| Lysosomal enzymes | Neutrophils, macrophages | Tissue degradation, microbial killing |
The arachidonic acid cascade is central: membrane phospholipids → arachidonic acid (via phospholipase A₂) → COX pathway (prostaglandins, thromboxane) or LOX pathway (leukotrienes). NSAIDs inhibit COX; corticosteroids inhibit phospholipase A₂ (upstream of both).
5. Chronic Inflammation
5.1 Definition
Chronic inflammation is a prolonged response (weeks to months) in which inflammation, tissue injury, and attempts at repair coexist. It may arise:
- Following unresolved acute inflammation
- De novo as a smoldering process
5.2 Causes
- Persistent infections (mycobacteria, certain fungi, viruses, parasites)
- Hypersensitivity/autoimmune diseases (rheumatoid arthritis, multiple sclerosis, asthma)
- Prolonged toxic exposure — inhaled silica (silicosis), cholesterol/lipids (atherosclerosis)
5.3 Morphologic Features
Three hallmarks:
- Infiltration with mononuclear cells — macrophages, lymphocytes, plasma cells
- Tissue destruction (induced by the inflammatory cells)
- Attempts at repair — angiogenesis + connective tissue replacement (fibrosis)
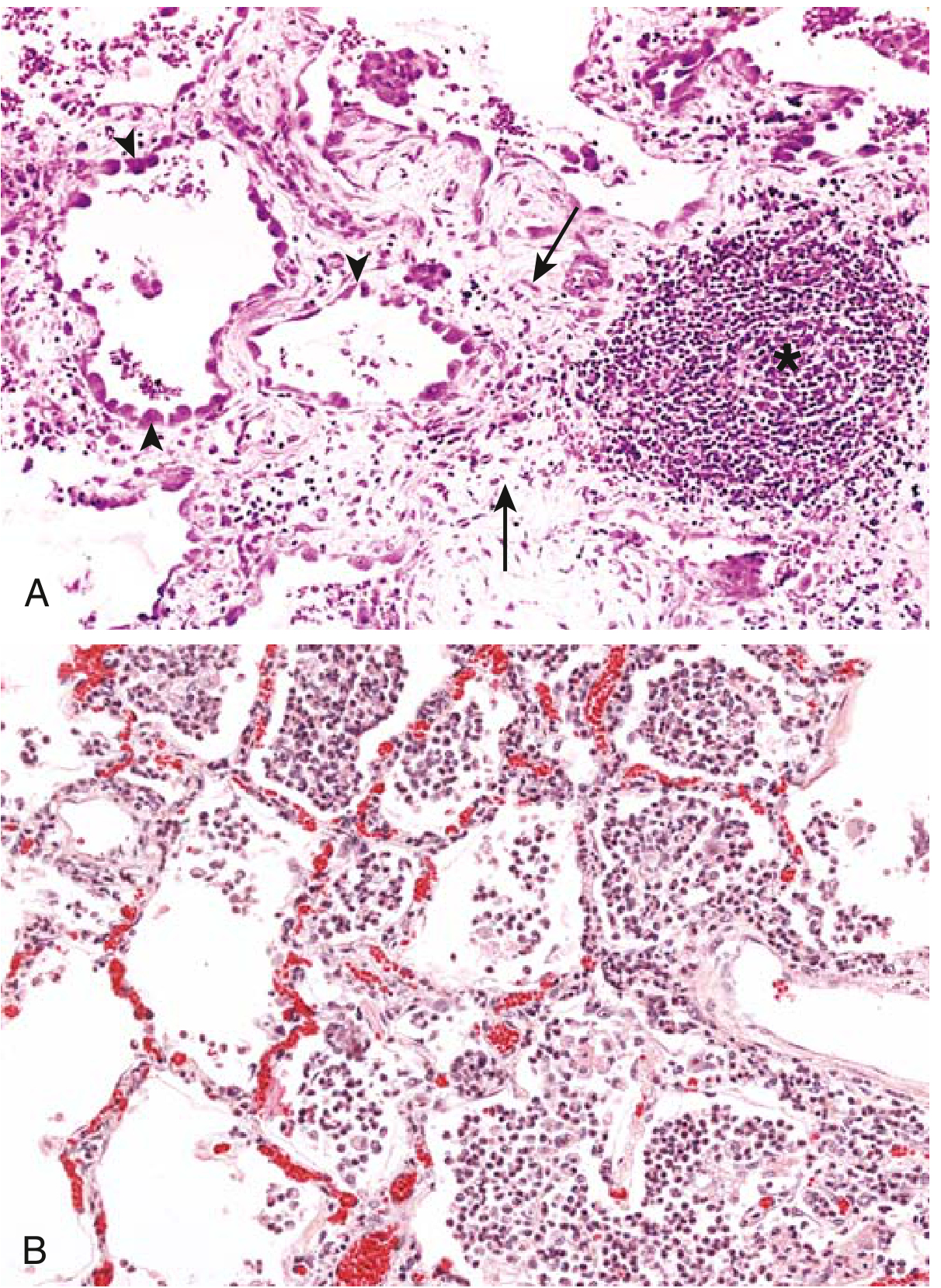
5.4 Key Cells in Chronic Inflammation
Macrophages (dominant cell)
- Derived from blood monocytes or tissue-resident populations (Kupffer cells, microglia, alveolar macrophages)
- Two polarization states:
- M1 (classical activation): Triggered by IFN-γ, microbial products → produces NO, ROS, pro-inflammatory cytokines (TNF, IL-1, IL-12) → kills microbes
- M2 (alternative activation): Triggered by IL-4, IL-13 (from Th2 cells) → promotes tissue repair, fibrosis, angiogenesis; secretes TGF-β, VEGF
Lymphocytes
- T helper 1 (Th1) cells → IFN-γ → activate macrophages
- Th2 cells → IL-4, IL-5, IL-13 → activate eosinophils, stimulate IgE (allergic reactions)
- Th17 cells → IL-17 → neutrophil recruitment, antimicrobial peptides
- B cells → mature to plasma cells → antibody secretion
Plasma Cells — produce antibodies directed at persistent antigens
Eosinophils — prominent in parasitic infections and allergies
Mast Cells — key in allergic reactions and surveillance
6. Granulomatous Inflammation
A specialized pattern of chronic inflammation characterized by aggregates of activated macrophages (epithelioid cells), often surrounded by lymphocytes.
- Formed when injurious agents (microbes, foreign bodies) cannot be eliminated by standard phagocytosis
- Giant cells may form by macrophage fusion (Langerhans cells in TB, foreign-body giant cells)
- Central caseous necrosis = soft, cheese-like necrosis (characteristic of tuberculosis)
| Disease | Cause | Granuloma type |
|---|---|---|
| Tuberculosis | M. tuberculosis | Caseating; acid-fast bacilli; Langhans giant cells |
| Leprosy | M. leprae | Non-caseating; acid-fast bacilli in macrophages |
| Sarcoidosis | Unknown | Non-caseating; abundant epithelioid macrophages |
| Crohn disease | Immune/bacterial | Non-caseating; in intestinal wall |
| Syphilis | T. pallidum | Gumma; plasma cell infiltrate; central necrosis |
| Cat-scratch disease | Bartonella henselae | Rounded/stellate with neutrophils; giant cells uncommon |
7. Systemic Effects of Inflammation — Acute Phase Response
When inflammation is severe or systemic, it triggers the acute-phase response, mediated primarily by IL-1, TNF-α, and IL-6:
| Manifestation | Mechanism |
|---|---|
| Fever | Cytokines (IL-1, TNF, IL-6) → hypothalamic PGE₂ → raised thermostat setpoint |
| Leukocytosis | IL-1, TNF → ↑ marrow production; shift left (band forms) in bacterial infections |
| ↑ CRP, fibrinogen, SAA | IL-6 → hepatocyte synthesis; CRP acts as opsonin; SAA replaces HDL apolipoproteins |
| ↑ ESR | Fibrinogen causes rouleaux formation → faster red cell sedimentation |
| Anemia of chronic disease | IL-6 → ↑ hepcidin → reduced iron availability |
| Hypotension, DIC (septic shock) | Massive TNF, IL-1, IL-6 from widespread infection → endothelial/coagulation activation |
8. Anti-Inflammatory Drug Targets (Clinical Correlations)
| Drug class | Target | Effect |
|---|---|---|
| NSAIDs (aspirin, ibuprofen) | COX-1/COX-2 | ↓ Prostaglandins → ↓ fever, pain, vascular permeability |
| Corticosteroids | Phospholipase A₂ (via lipocortin); NF-κB | ↓ All arachidonic acid derivatives; suppress cytokines |
| Antihistamines | H₁ receptor | Block histamine-mediated vasodilation/permeability |
| Anti-TNF biologics (etanercept, infliximab) | TNF-α | Reduce chronic inflammatory diseases (RA, IBD) |
| IL-1 antagonists (anakinra) | IL-1 receptor | Block IL-1–mediated systemic effects |
| Colchicine | Tubulin (microtubules) | ↓ Neutrophil chemotaxis and NET formation (gout) |
9. Summary Flow
Injurious stimulus (infection, trauma, necrosis)
↓
Recognition by PRRs (TLRs, NLRs) on tissue macrophages
↓
Release of vasoactive amines (histamine) + cytokines (TNF, IL-1)
↓
VASCULAR CHANGES: vasodilation → ↑ permeability → exudation
↓
CELLULAR CHANGES: margination → rolling (selectins) →
firm adhesion (integrins/ICAM) → transmigration (PECAM-1) →
chemotaxis → phagocytosis + killing (ROS, MPO, enzymes)
↓
┌──────────────┐
↓ ↓
RESOLUTION CHRONIC INFLAMMATION
(macrophage (macrophages + lymphocytes
clearance) + tissue damage + fibrosis)
↓
GRANULOMA (if agent
cannot be cleared)
Sources:
- Robbins & Kumar Basic Pathology, 10th ed. — Chapter 2 (Inflammation and Repair), pp. 44–70
- Robbins, Cotran & Kumar Pathologic Basis of Disease — Chapter 3
This is a shared conversation. Sign in to Orris to start your own chat.