Test for genetic cause of hyper triglycerides
Reading File
Finding Sources
Finding Sources
Searching PubMed
hypertriglyceridemia genetic testing diagnosis
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading Article
PMID: 41472374
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
I now have everything needed for a thorough answer. Here is the complete synthesis:
Genetic Testing for Hypertriglyceridemia
Background: Why Genetics Matter
Hypertriglyceridemia (HTG) has a strong genetic basis. Triglyceride (TG) levels across a spectrum are shaped by both common low-effect variants (polygenic) and rare large-effect single-gene (monogenic) mutations. Understanding this distinction determines which test to order and when.
- TG 150-899 mg/dL: usually polygenic - many common and rare variants with small individual effects
- TG >900 mg/dL: strongly suggests a large-effect monogenic disorder
- TG >1000 mg/dL: risk of pancreatitis rises sharply; lifetime risk ~5-20% depending on severity
(Goldman-Cecil Medicine, p. 2290; Sleisenger & Fordtran, p. 1035)
Genetic Disorders Causing Hypertriglyceridemia
1. Familial Chylomicronemia Syndrome (FCS) - Monogenic, Autosomal Recessive
The "gold standard" for genetic HTG. Caused by complete loss-of-function mutations in genes encoding the LPL pathway:
| Gene | Protein | Inheritance | Mechanism |
|---|---|---|---|
| LPL | Lipoprotein lipase | AR | Cannot hydrolyze chylomicrons; most common cause |
| APOC2 | Apolipoprotein C-II | AR | Loss of LPL activator (functional LPL deficiency) |
| APOA5 | Apolipoprotein A-V | AR | Cofactor for LPL activation lost |
| GPIHBP1 | GPI-HDL binding protein 1 | AR | Anchors LPL to endothelium; loss impairs lipolysis |
| LMF1 | Lipase maturation factor 1 | AR | Required for LPL folding |
Clinically: presents in childhood with recurrent abdominal pain, acute pancreatitis, fasting TG often >1000-10,000 mg/dL, eruptive xanthomas, hepatosplenomegaly, lipemia retinalis. FCS is rare (~1-2 in 1,000,000).
(Henry's Clinical Diagnosis, p. 3140; Sleisenger & Fordtran, p. 1035)
2. Multifactorial Chylomicronemia Syndrome (MCS) - Complex/Polygenic
The most common cause of severe HTG in adults. Not a single-gene disorder - patients carry a combination of:
- Heterozygous loss-of-function mutations in TG-raising genes (LPL, APOA5, etc.)
- Common pathogenic variants at multiple loci
- Secondary factors: obesity, diabetes, alcohol, medications
An expert panel proposed the FCS Score to help distinguish FCS from MCS (important because emerging RNA therapies are targeted specifically at FCS). However, this has not been fully validated.
(Sleisenger & Fordtran, p. 1035)
3. Familial Hypertriglyceridemia (Type IV) - Autosomal Dominant
- Population prevalence 5-10%
- Isolated VLDL elevation, TG usually 200-500 mg/dL
- Pathophysiology: overproduction of triglyceride-rich VLDL with normal apoB
- Specific causative gene(s) remain incompletely defined; often exacerbated by obesity and insulin resistance
(Henry's Clinical Diagnosis, p. 3133)
4. Familial Combined Hyperlipidemia (Type IIB)
- Most common primary hyperlipoproteinemia (~1 in 100)
- Elevated LDL + TG (mixed pattern)
- Phenotypic heterogeneity within families
- No single definitive biochemical or genetic marker; diagnosis requires family history showing multiple lipid phenotypes
5. Dysbetalipoproteinemia (Type III) - APOE-related
- APOE e2/e2 genotype is the classic predisposing variant
- TG and cholesterol both elevated; characteristic broad-beta band on electrophoresis
- VLDL-C:TG ratio >0.3 (vs <0.2 in normal) is diagnostic
- APOE genotyping is the confirmatory genetic test
6. ApoC-III Excess
- Multiple polymorphisms in APOC3 gene (e.g., -455T/C, -482C/T, 1100C/T)
- ApoC-III inhibits LPL activity and promotes VLDL production
- Contributes to elevated TG, especially in metabolic syndrome and T2DM
When to Order Genetic Testing
Genetic testing is selectively used, not routine. Per the 2026 Hegele review in JCEM (PMID 41472374), the key indications are:
| Clinical Scenario | Recommended Test |
|---|---|
| TG >885 mg/dL (≥10 mmol/L), childhood onset, no secondary cause | FCS gene panel (LPL, APOC2, APOA5, GPIHBP1, LMF1) |
| Suspected dysbetalipoproteinemia (broad-beta band, mixed hyperlipidemia) | APOE genotype |
| Beta-sitosterolemia (high cholesterol + xanthomas, low LDL-C on treatment) | ABCG5/ABCG8 sequencing |
| Family history of severe HTG with pancreatitis | FCS gene panel |
| Distinguishing FCS from MCS (eligibility for RNA-based therapy) | FCS gene panel |
| Mild-moderate HTG (150-500 mg/dL) | Genetic testing NOT routinely indicated |
Practical Testing Approach
Step 1 - Fasting lipid panel + secondary cause workup first
Rule out: uncontrolled diabetes, hypothyroidism, pregnancy, renal disease, obesity, alcohol, offending drugs (estrogens, glucocorticoids, protease inhibitors, thiazides, retinoids).
Step 2 - Lipoprotein phenotyping
- Lipoprotein electrophoresis (agarose gel): identifies Fredrickson type (I, IIB, III, IV, V)
- Panel C below shows chylomicronemia at origin (TG = 3840 mg/dL); Panel B shows type III beta-VLDL pattern
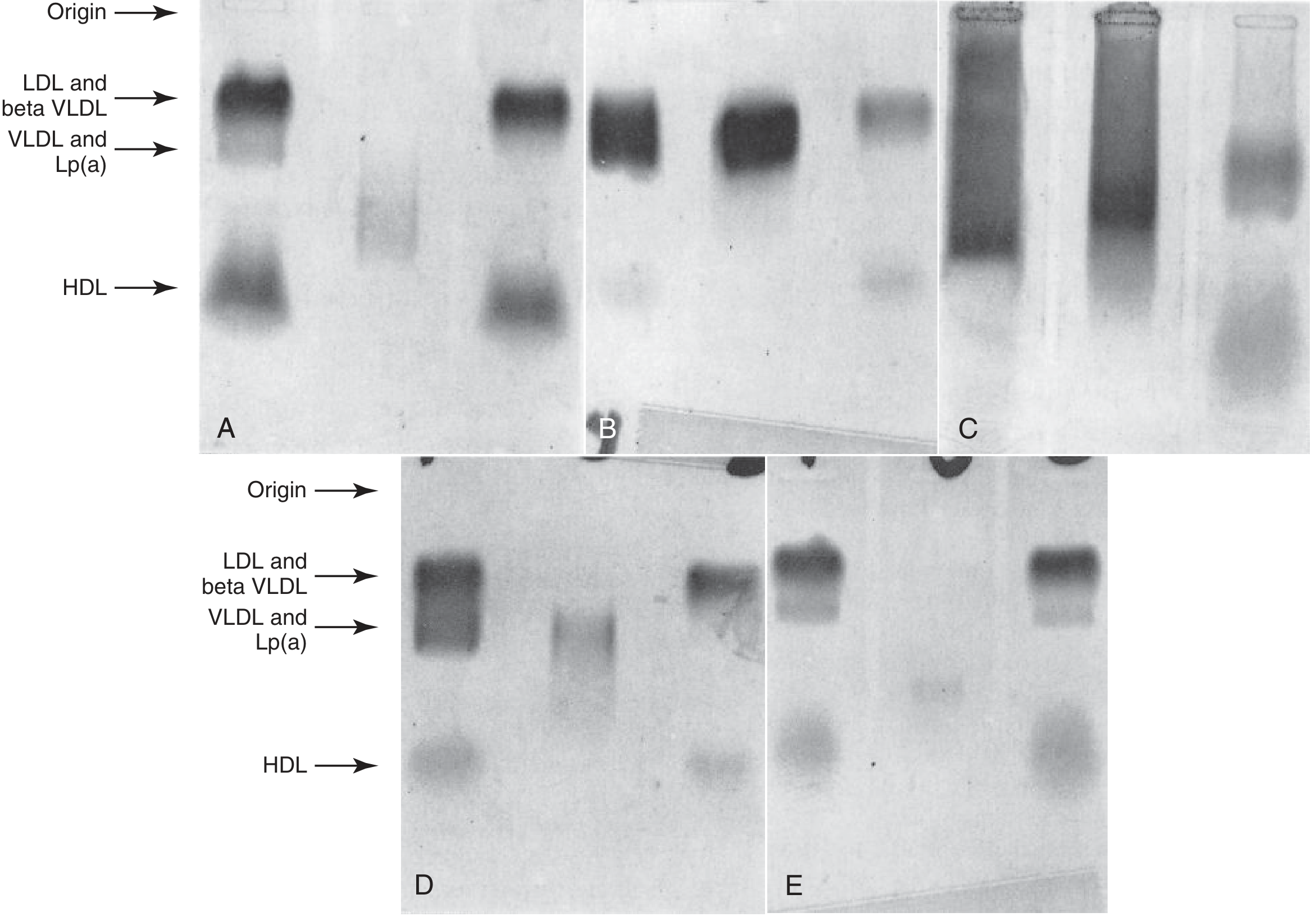
Step 3 - Targeted genetic testing when indicated
- FCS gene panel: sequences LPL, APOC2, APOA5, GPIHBP1, LMF1 for pathogenic variants
- APOE genotyping: for suspected type III
- ABCG5/ABCG8: for suspected beta-sitosterolemia
- ApoB measurement and TG:total cholesterol ratio help triage
Step 4 - Family screening
When severe HTG (>500 mg/dL) is confirmed in a proband, screen first-degree relatives (screening and counseling is recommended given clustering due to shared genetic + environmental factors).
Why It Matters Clinically (2026 Update)
The Hegele 2026 review (PMID 41472374) emphasizes: the overwhelming majority of adults presenting with chylomicronemia have MCS, not FCS. However, correctly identifying FCS is now therapeutically critical because RNA-based therapies targeting apolipoprotein C-III - volanesorsen, olezarsen, and plozasiran - are approved or emerging specifically for FCS and refractory MCS. These drugs require genetic confirmation for proper patient selection.
Also note: the 2024 review on clinical impact of genetic testing for lipid disorders (PMID 38456469) confirms genetic testing is reshaping dyslipidemia management, with clinical utility extending beyond FH to include hypertriglyceridemia syndromes.
Summary Table
| Disorder | Fredrickson | Key Gene(s) | Test |
|---|---|---|---|
| Familial chylomicronemia (FCS) | Type I | LPL, APOC2, APOA5, GPIHBP1, LMF1 | Gene panel |
| Familial hypertriglyceridemia | Type IV | Unknown (polygenic-dominant) | Clinical/family Hx |
| Familial combined hyperlipidemia | Type IIB | Unknown (polygenic) | Clinical/family Hx |
| Dysbetalipoproteinemia | Type III | APOE (e2/e2) | APOE genotype |
| Multifactorial chylomicronemia (MCS) | Type V | Multiple + secondary | FCS panel to exclude FCS |
| Beta-sitosterolemia | - | ABCG5, ABCG8 | Sitosterol level + gene sequencing |
Sources: Henry's Clinical Diagnosis and Management by Laboratory Methods, p. 3133-3158 | Goldman-Cecil Medicine, p. 2290 | Sleisenger & Fordtran's GI and Liver Disease, p. 1035 | Tietz Textbook of Laboratory Medicine, p. 594-602 | Hegele RA, J Clin Endocrinol Metab 2026 (PMID 41472374)
Cmv retinitis
Reading File
Finding Sources
Finding Sources
Searching PubMed
cytomegalovirus retinitis treatment
Reading File
Finding Sources
Finding Sources
Reading Article
PMID: 39339903
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the material needed for a comprehensive answer. Here is the full synthesis:
CMV Retinitis
Overview
Cytomegalovirus (CMV) retinitis is the most common serious ocular opportunistic infection in immunocompromised patients, accounting for 85% of CMV end-organ disease in AIDS. It typically occurs with profound CD4 depletion (CD4 <50 cells/μL). CMV is a herpesvirus - latent in most adults, reactivating when cell-mediated immunity collapses. Without treatment, severe visual loss is essentially inevitable. Since the advent of antiretroviral therapy (ART), incidence has fallen substantially, though prevalence remains significant.
Epidemiology & Risk Factors
- CD4 count <50 cells/μL: highest risk (screening every 3 months)
- CD4 50-100 cells/μL: screen every 6 months
- CD4 >100 cells/μL: yearly screening
- HIV microangiopathy (cotton-wool spots) is a marker for increased CMV retinitis risk
- Also occurs in: bone marrow and solid organ transplant recipients, patients on chronic immunosuppressive therapy, congenital CMV
- Risk is higher in patients on systemic corticosteroids even after ART
Symptoms
- Often asymptomatic early (especially peripheral lesions)
- Floaters
- Scotomata (focal visual field defects)
- Peripheral visual field loss
- Reduced central acuity when macula is involved
- Photopsia
Fundoscopic Appearance
The hallmark is a full-thickness necrotizing retinitis. Two recognized clinical patterns:
Fulminant (edematous) pattern - starts near the posterior pole:
- Dense white/yellow areas of retinal infiltration and necrosis
- Prominent flame-shaped intraretinal hemorrhages
- Classic "pizza pie" or "Margherita pizza" appearance - white necrosis with bright red hemorrhage interspersed
Indolent (granular) pattern - peripheral, less aggressive:
- Granular white infiltrates with fewer hemorrhages
- Less vasculitis
- Can spread silently toward the macula over weeks
Other features:
- Retinal vasculitis (vascular sheathing)
- Frosted branch angiitis - marked vascular sheathing in ~6%
- Optic neuritis from direct spread
- Mild anterior uveitis
- Minimal vitreous inflammation (unlike acute retinal necrosis)
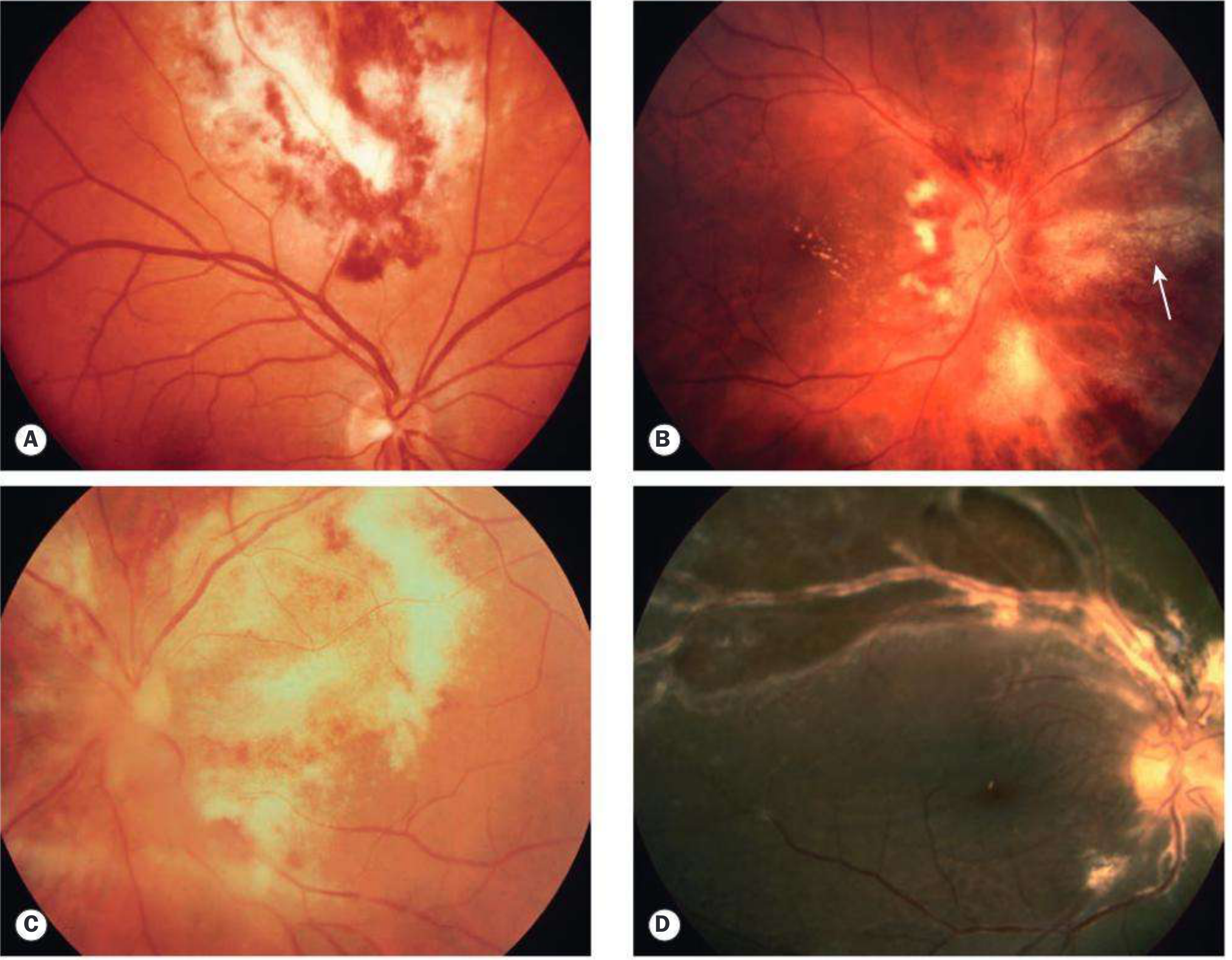
Kanski's Clinical Ophthalmology, Fig. 12.42 - CMV retinitis spectrum
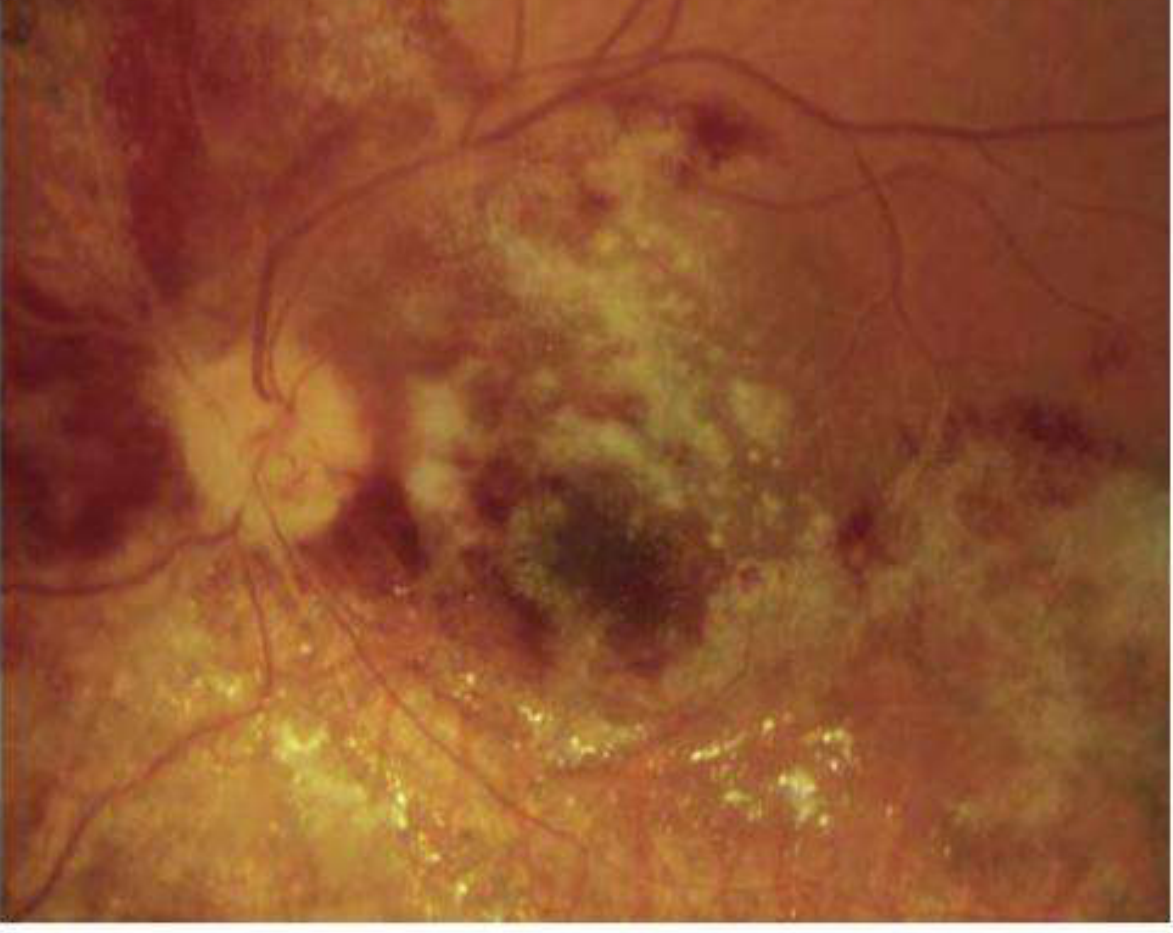
Kanski's Clinical Ophthalmology, Fig. 12.43 - Regressing CMV retinitis post-treatment
Complications
| Complication | Notes |
|---|---|
| Retinal detachment | Necrotic retina leaves holes/tears; occurs in up to 50%; requires vitrectomy + silicone oil |
| Progressive contralateral spread | Both eyes affected in 50% if untreated |
| Optic atrophy | From optic nerve involvement |
| Cataract | Common late-stage finding |
| Immune Recovery Uveitis (IRU) | See below |
Diagnosis
- Clinical: ophthalmoscopy showing characteristic fundus appearance is the primary diagnostic method
- CMV PCR (blood/vitreous): viremia usually present with active end-organ disease; supports diagnosis but can be present without retinitis
- CD4 count
- No biopsy needed in typical cases
- Differential: toxoplasma retinochoroiditis, progressive outer retinal necrosis (PORN/VZV), acute retinal necrosis, syphilis, HIV retinopathy, intraocular lymphoma
Treatment
Treatment has two phases: induction followed by maintenance.
First-Line: Valganciclovir (preferred - oral bioavailability = IV ganciclovir)
| Phase | Dose | Duration |
|---|---|---|
| Induction | 900 mg PO twice daily | 14-21 days |
| Maintenance | 900 mg PO once daily | Indefinite until immune recovery |
Discontinue when CD4 >100-150 cells/μL sustained on ART.
IV Ganciclovir (when PO not feasible)
| Phase | Dose |
|---|---|
| Induction | 5 mg/kg IV twice daily × 14-21 days |
| Maintenance | 5 mg/kg IV once daily |
- Main toxicity: myelosuppression/neutropenia - treat with G-CSF (filgrastim)
Alternative Systemic Agents (sight-threatening lesions, ganciclovir resistance, intolerance)
| Drug | Route | Key Toxicity | Notes |
|---|---|---|---|
| Foscarnet | IV | Nephrotoxicity, electrolyte abnormalities (Ca, Mg, K) | Best CNS penetration; use for CMV encephalitis; hydrate well |
| Cidofovir | IV | Nephrotoxicity, uveitis, decreased IOP | Give probenecid + IV saline to reduce nephrotoxicity |
Local/Intravitreal Therapy (adjunct for sight-threatening lesions near optic nerve or macula)
- Intravitreal ganciclovir 2 mg/injection
- Intravitreal foscarnet 1.2 mg
- Intravitreal cidofovir 20 μg
- Note: ganciclovir implant marketing authorization has been withdrawn
ART (Cornerstone)
- ART is the mainstay of long-term management - restores immune function and CMV control
- Start ART promptly (as early as tolerated)
Drug Resistance
- UL97 kinase mutations: confer resistance to ganciclovir/valganciclovir; usually remain sensitive to foscarnet and cidofovir
- UL54 DNA polymerase mutations (in addition to UL97): confer resistance to ganciclovir, foscarnet, AND cidofovir
- Letermovir: novel mechanism (CMV terminase complex inhibitor); active against ganciclovir-resistant strains; used for prophylaxis in transplant recipients
Immune Recovery Uveitis (IRU)
A significant late complication after starting ART:
- Cause: Rejuvenated immune response against residual CMV antigen after CD4 recovery
- Features: Marked vitritis, cystoid macular edema, epiretinal membrane, cataract - can progress to phthisis bulbi
- Treatment: Intravitreal or systemic corticosteroids (with caution)
- Paradoxically, patients with best immune recovery have highest IRU risk
Differential Diagnosis Summary
| Condition | Key Distinguishing Feature |
|---|---|
| CMV retinitis | Dense white necrosis + hemorrhage ("pizza pie"), CD4 <50, minimal vitritis |
| Toxoplasma | "Headlight in fog" - focal retinochoroiditis with heavy vitritis, old scar |
| PORN (VZV) | CD4 <50, rapid necrosis, minimal vitritis, no hemorrhage, multifocal white-yellow lesions |
| Acute retinal necrosis | Immunocompetent, severe vitritis, peripheral necrosis progresses rapidly |
| Syphilitic retinitis | Can mimic CMV; positive VDRL/RPR |
| HIV microangiopathy | Cotton-wool spots only, asymptomatic, resolve spontaneously |
Monitoring
- Ophthalmology review every 2 weeks during active disease/induction
- Once stable on maintenance: monthly to 3-monthly
- Fundus photography and OCT useful for monitoring lesion borders and macular involvement
Sources: Kanski's Clinical Ophthalmology 10th ed., p. 489-490 | Washington Manual of Medical Therapeutics, p. 643 | Harrison's Principles of Internal Medicine 22e, p. 1529 | Henry's Clinical Diagnosis and Management, p. 2030 | Goodman & Gilman's Pharmacological Basis of Therapeutics | Zhang et al., Viruses 2024 (PMID 39339903) | Kobayashi & Hashida, Viruses 2024 (PMID 39066272)
Cause of blepharospasm
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive content from four authoritative sources. Here is the complete answer:
Causes of Blepharospasm
Blepharospasm is bilateral, repetitive, involuntary contraction of the orbicularis oculi muscles, leading to forced eyelid closure. It is classified into two broad categories: primary (essential/idiopathic) and secondary (reflex or symptomatic).
1. Primary (Essential / Idiopathic) Blepharospasm
The most common form. Also called benign essential blepharospasm (BEB).
- Etiology is idiopathic and likely multifactorial, possibly involving dysfunction of dopaminergic pathways within the basal ganglia
- Neuroimaging studies (PET-FDG, fMRI) show abnormal activity in: primary motor cortex, anterior cingulate gyrus, supplementary motor area, striatum, thalamus, inferior frontal lobes, pons, and cerebellum
- Pathologic changes have been identified in the primary motor cortex, anterior cingulate, and premotor/supplementary motor cortices
- No consistent neuropathologic lesion or neurochemical profile has been established
- Genetic basis is possible but few cases are clearly inherited; no association with known dystonia genes confirmed
- Predominantly women, onset in the sixth decade
- When combined with oromandibular dystonia (jaw, tongue spasms) = Meige syndrome (also called Brueghel syndrome)
- May also be associated with spasmodic dysphonia, torticollis, and other dystonic fragments
2. Secondary / Symptomatic Causes
A. Ocular Surface Disease (Reflex Blepharospasm)
The most common secondary cause - local irritation triggering protective reflexes:
- Dry eye syndrome
- Blepharitis (including rosacea of the eyelids)
- Corneal or conjunctival foreign body
- Trichiasis (misdirected eyelashes)
- Keratitis / corneal ulcer
- Iritis / anterior uveitis - severe reflex blepharospasm
- Conjunctivitis
Important: Spasms from ocular inflammation persist even after topical anaesthetic application - a key clinical differentiator from purely reflex blepharospasm.
B. Drug-Induced (Tardive Dystonia)
- Antipsychotics (phenothiazines, butyrophenones) - tardive dyskinesia / tardive dystonia
- Neuroleptics - long-term use
- Dopamine antagonists
- Blepharospasm is a recognized form of medication-induced acute or tardive dystonia
C. Neurological / Basal Ganglia Disorders
| Condition | Notes |
|---|---|
| Parkinson disease | Light closure of eyelids may induce blepharospasm; also associated with apraxia of eyelid opening |
| Progressive supranuclear palsy (PSP) | Rostral brainstem disease disrupts dopaminergic/cholinergic pathways |
| Wilson disease | Basal ganglia involvement |
| Huntington disease | Caudate nucleus dysfunction |
| Multiple system atrophy (MSA-C) | Rostral brainstem lesions causing disinhibition of brainstem reflexes |
| Pantothenate kinase-associated neurodegeneration (PKAN) | Globus pallidus iron accumulation |
| Postencephalitic disorders | Residual basal ganglia damage |
D. Cerebrovascular Disease
- Cerebral infarction or hemorrhage - especially in the days following stroke; more commonly with left hemisphere (non-dominant) lesions
- Thalamomesencephalic infarct - homolateral blepharospasm
- Bilateral infarctions of rostral brainstem, diencephalon, or striatum
- Left frontal cortical infarction - asymmetric blepharospasm
- Non-dominant striatal infarctions - bilateral transient blepharospasm
E. Demyelinating Disease
- Multiple sclerosis - brainstem plaques involving the rostral brainstem
F. Other Neurological/Systemic Causes
- Retinal degeneration
- Familial apoceruloplasmin deficiency
- Familial syndrome with dystonia and pigmentary retinopathy
- Paraneoplastic midbrain encephalitis
- Autoimmune disease (e.g., systemic lupus erythematosus)
- Myasthenia gravis (can mimic essential blepharospasm through lid fatigue)
G. Functional (Psychogenic)
- Rarely, blepharospasm has a functional basis - eye closure with lid fluttering in suggestible patients
- Psychiatric causation is not supported for essential blepharospasm - psychotherapy and biofeedback have failed to help
- Functional cases are distinguished by suggestibility and inconsistency of findings
Key Distinguishing Features: Blepharospasm vs. Mimics
| Condition | Laterality | During Sleep | Other Clues |
|---|---|---|---|
| Essential blepharospasm | Bilateral | Disappears | Women >60, worsened by stress/light |
| Hemifacial spasm | Unilateral | Persists | Entire face, CN VII compression |
| Eyelid myokymia | Unilateral (lower lid) | Self-limited | Stress, caffeine, fatigue |
| Apraxia of eyelid opening | Bilateral | - | No orbicularis spasm; common in Parkinson disease |
| Tardive dyskinesia | Bilateral | - | Drug history (antipsychotics) |
| Tourette syndrome | Variable | - | Multiple tics, vocalisations |
| Reflex (ocular) | Bilateral | - | Ocular surface pathology, photophobia |
Precipitating / Aggravating Factors (for essential blepharospasm)
- Bright light (photophobia very common)
- Stress
- Reading and watching television
- Relieved by: talking, relaxation, touching the face, singing, and sleep
Sources: Adams & Victor's Principles of Neurology 12th ed., p. 111-112 | Localization in Clinical Neurology 8th ed., p. 799 | Wills Eye Manual, p. 393-394 | Kanski's Clinical Ophthalmology 10th ed., p. 854
obesity & recent advances in management
Finding Sources
Searching PubMed
obesity pharmacotherapy GLP-1 management
Reading File
Finding Sources
Reading Article
PMID: 39952695
Reading File
Reading File
Reading Article
PMID: 39037780
Reading File
Reading File
Searching PubMed
retatrutide orforglipron obesity phase 3 trial
Finding Sources
Finding Sources
Reading File
I have extensive material from multiple top-tier sources. Let me now compile the comprehensive answer.
Obesity and Recent Advances in Management
Definition & Classification
Obesity is defined as excess body fat sufficient to impair health. The standard classification uses Body Mass Index (BMI = weight kg / height m²):
| Classification | BMI (kg/m²) |
|---|---|
| Overweight | 25.0 - 29.9 |
| Obesity Class I | 30.0 - 34.9 |
| Obesity Class II | 35.0 - 39.9 |
| Obesity Class III (severe) | ≥ 40 |
Waist circumference is an important additional metric - abdominal/central obesity (>102 cm men, >88 cm women) confers independent cardiometabolic risk beyond BMI.
Pathophysiology
Energy Imbalance & Adipose Dysfunction
Obesity results from chronic caloric excess relative to expenditure, but the underlying biology is complex:
- Free fatty acid (FFA) overabundance: Expanded adipose tissue releases FFA excessively → FFA impairs insulin-mediated glucose uptake in muscle, increases hepatic glucose and triglyceride production, and drives VLDL secretion
- Leptin resistance: In obesity, leptin (the adipokine that suppresses appetite and promotes energy expenditure) loses its signaling efficacy ("leptin resistance") - so the normal appetite-braking mechanism fails
- Insulin resistance: Central to metabolic syndrome. The inhibition of lipolysis by insulin is the most sensitive pathway - once this fails, a vicious cycle of lipolysis → FFA excess → worsening insulin resistance ensues
Hormonal Regulation of Appetite
Key gut-brain axis hormones:
- GLP-1 (glucagon-like peptide-1): incretin released from intestinal L-cells; suppresses appetite via hypothalamus, slows gastric emptying, stimulates insulin, inhibits glucagon
- GIP (glucose-dependent insulinotropic polypeptide): synergizes with GLP-1 for insulin release and adipose metabolism
- Amylin / PYY / CCK: satiety hormones reduced or dysfunctional in obesity
- Ghrelin: "hunger hormone" from stomach - elevated in states of caloric restriction, blunted after surgery
Complications of Obesity
- Cardiovascular: coronary artery disease, heart failure (HFpEF), hypertension, stroke (2-3× risk)
- Metabolic: type 2 diabetes, dyslipidemia, metabolic syndrome, NAFLD/MASLD
- Respiratory: obstructive sleep apnea, obesity hypoventilation syndrome
- Musculoskeletal: osteoarthritis
- Cancer: endometrial, breast, colorectal, esophageal, pancreatic, kidney
- Psychosocial: depression, reduced quality of life
Management Framework: Two Approaches
1. BMI-Centric (NHLBI) Stepwise Model
| BMI | Lifestyle | Pharmacotherapy | Surgery |
|---|---|---|---|
| 25-26.9 | Yes | No | No |
| 27-29.9 | Yes | With comorbidities | No |
| 30-34.9 | Yes | Yes | No |
| 35-39.9 | Yes | Yes | With comorbidities |
| ≥40 | Yes | Yes | Yes |
2. Complications-Centric (AACE) Model
Focuses on treating obesity-related complications rather than just lowering BMI:
- Stage 0 (no complications): prevent weight gain; lifestyle only
- Stage 1 (mild-moderate complications): lifestyle + obesity medication
- Stage 2 (serious complications): lifestyle + medication + consider bariatric surgery
Pillar 1: Lifestyle Intervention
Foundation of all treatment - must accompany any pharmacotherapy or surgery:
- Caloric deficit of 500-750 kcal/day
- High-intensity comprehensive lifestyle intervention (behavioral counseling, dietary change, physical activity)
- Goal: ≥5% weight loss to improve metabolic parameters; ≥10% for more significant comorbidity improvement
- Physical activity: ≥150 min/week moderate intensity; ≥200-300 min/week for weight maintenance
Pillar 2: Pharmacotherapy
FDA-Approved Anti-Obesity Medications (AOMs)
Comparative weight loss vs. placebo (meta-analysis, Gudzune & Kushner, JAMA 2024):
| Drug | Mechanism | Weight Loss vs Placebo | Key Side Effects |
|---|---|---|---|
| Orlistat (Xenical) | Pancreatic lipase inhibitor (GI fat absorption ↓) | −3.1% | Oily spotting, fecal urgency, fat-soluble vitamin ↓ |
| Phentermine (short-term) | Sympathomimetic, appetite suppressant | Variable | Tachycardia, hypertension (short-term only, 3 months) |
| Phentermine/Topiramate ER | Sympathomimetic + GABA modulation | −8.0% | Constipation, paresthesia, cognitive effects, teratogenic |
| Naltrexone/Bupropion | Opioid antagonist + dopamine/noradrenaline reuptake inhibitor | −4.1% | Nausea, constipation; CV risk uncertain |
| Liraglutide 3 mg | GLP-1 receptor agonist | −4.7% | Nausea, vomiting, pancreatitis risk |
| Semaglutide 2.4 mg | GLP-1 receptor agonist | −11.4% | Nausea (28-44%), diarrhea, constipation |
| Tirzepatide 15 mg | Dual GIP + GLP-1 receptor agonist | −12.4 to −19.2% | Nausea, vomiting, diarrhea |
Pillar 3: Bariatric / Metabolic Surgery
Indications:
- BMI ≥40, OR
- BMI 35-40 with serious comorbidity (especially uncontrolled T2DM), after failed medical management
- Some guidelines now consider BMI ≥35 with or without comorbidities
Types and outcomes:
| Procedure | Mechanism | Weight Loss | Notes |
|---|---|---|---|
| Roux-en-Y Gastric Bypass (RYGB) | Restriction + mild malabsorption + gut hormone changes | 25-35% of body weight | Gold standard; best for T2DM remission |
| Laparoscopic Sleeve Gastrectomy (LSG) | Restriction + ghrelin reduction | ~25-30%; nearly equivalent to RYGB | Most commonly performed; fewer complications |
| Adjustable Gastric Band | Restriction only | Inferior long-term results | Rarely used now; high complication rate |
| Biliopancreatic Diversion/Duodenal Switch | Restriction + malabsorption | Greatest weight loss | Highest risk for nutritional deficiencies |
Benefits beyond weight loss:
- T2DM remission in 29-43% after RYGB vs. 5-7% with medical therapy alone (ARMMs-T2D study)
- Retinopathy risk reduced by 71%, ESRD by 69%, nephropathy by 59%
- Reduction in hypertension, dyslipidemia, cardiovascular mortality, and obesity-associated cancers
- ~70% of patients achieve success; most weight loss in first 1-2 years
- Operative mortality <0.5% at expert centers
Recent Advances (2023-2026)
1. Semaglutide (Ozempic/Wegovy) - The Benchmark Shift
- Weekly subcutaneous 2.4 mg: 14.9% average weight loss over 68 weeks (STEP trials)
- SELECT trial (2023): In people with obesity and established CVD (no diabetes), semaglutide reduced major adverse cardiovascular events (MACE) by 20% - the first anti-obesity drug to show cardiovascular mortality benefit
- This positioned GLP-1 RAs as disease-modifying rather than just weight-loss agents
2. Tirzepatide (Mounjaro/Zepbound) - Dual Incretin Agonist
- GIP + GLP-1 dual receptor agonist (once weekly SC injection)
- SURMOUNT trials: Mean weight loss of -22.5% at 72 weeks (15 mg dose) in people without diabetes - the largest drug-induced weight loss ever recorded in a phase 3 trial
- FDA-approved for obesity (2023)
- Placebo-subtracted difference: −19.2% vs. −12.9% with semaglutide in meta-analysis (Sabiston Textbook, 2024)
- Weight regain of ~two-thirds within 1 year if stopped - indicating need for chronic therapy
3. Oral Semaglutide 50 mg
- The only oral GLP-1 RA to complete a phase 3 trial (OASIS 1): ~15% weight loss
- Eliminates need for injections - major adherence advantage
- Phase 3 data now available; awaiting broader regulatory approval for obesity indication
4. Pipeline: Triple Agonists and Next-Generation Agents
From the Kokkorakis et al. systematic review, Pharmacol Rev 2025 (PMID 39952695) - 53 phase 2/3 trials identified, 36 emerging drugs:
| Drug | Mechanism | Phase | Weight Loss (Phase 2) |
|---|---|---|---|
| Retatrutide | GLP-1 + GIP + Glucagon triple agonist | Phase 3 ongoing | ~24% in phase 2 |
| CagriSema | GLP-1 RA + Amylin analog (cagrilintide) combination | Phase 3 ongoing | ~15-22% |
| Orforglipron | Oral small-molecule GLP-1 RA (non-peptide) | Phase 3 ongoing | ~14-15% |
| Mazdutide / Survodutide | GLP-1 + Glucagon dual agonist | Phase 3 ongoing | ~10-18% |
| Ecnoglutide / TG103 | GLP-1 RA variants | Phase 3 ongoing | - |
| Setmelanotide (Imcivree) | MC4 receptor agonist | Approved (2020) | Genetic obesity (POMC/LEPR/PCSK1 deficiency) |
Almost half of all drugs in phase 2 are incretin analogs. Phase 2 completed incretin-based therapies show mean weight loss of 7.4% to 24.2%.
5. GLP-1 RAs in New Indications (2024-2026)
- Heart failure (HFpEF): Semaglutide improves functional capacity and QoL in HFpEF regardless of diabetes status (STEP-HFpEF trial)
- MASLD/NASH: Tirzepatide up to 20.9% weight change with NASH resolution in phase 3; semaglutide global phase 3 trial ongoing
- Obstructive sleep apnea: Tirzepatide reduces AHI by ~63% (SURMOUNT-OSA, 2024)
- Kidney disease: SELECT and FLOW trial data confirm GLP-1 RAs slow CKD progression
6. Endoscopic Approaches (Emerging)
- Intragastric balloons: 6-month placement, reasonable option for BMI >30; more weight loss than lifestyle alone
- Endoscopic sleeve gastroplasty: suturing stomach into sleeve shape endoscopically; ~15-17% weight loss
- Duodenojejunal bypass liners: impermeable liner excludes proximal small bowel; under investigation
- Aspiration therapy: data still insufficient for routine use per Goldman-Cecil
7. GLP-1 RA Use Perioperatively
- Post-bariatric patients with inadequate weight loss benefit from semaglutide (mean additional 10% weight loss at 12 months, >60% achieving >10%)
- GLP-1 RAs are now used to "bridge" high-BMI patients to surgery as well as adjuvant post-surgery therapy
Choosing Between Options
Per Goldman-Cecil Medicine:
"Physicians should consider their patients' comorbid conditions and preferences to tailor treatment for each individual."
| Priority | Drug of Choice |
|---|---|
| Maximum weight loss (injectable tolerated) | Tirzepatide ≥ Semaglutide |
| Established CVD | Semaglutide (SELECT trial: 20% MACE reduction) |
| T2DM + obesity | Tirzepatide (also treats diabetes) |
| Oral preference | Phentermine/topiramate (cheapest), oral semaglutide 50 mg (emerging) |
| Cost sensitivity | Phentermine/topiramate ER |
| Contraindications to GLP-1 | Naltrexone/bupropion or phentermine/topiramate |
| BMI ≥35 + serious comorbidity failing medical therapy | Bariatric surgery |
Key caveat: Weight regain after stopping AOMs approaches two-thirds of prior weight loss within 1 year, supporting the view that obesity requires chronic, continuous treatment - analogous to hypertension or diabetes.
Sources: Fuster & Hurst's The Heart 15th ed. | Goldman-Cecil Medicine | Sabiston Textbook of Surgery | Harrison's Principles 22nd ed. (2025) | Gudzune & Kushner, JAMA 2024 (PMID 39037780) | Kokkorakis et al., Pharmacol Rev 2025 (PMID 39952695) | Hamza et al., Expert Opin Pharmacother 2025 (PMID 39632534)
This is a shared conversation. Sign in to Orris to start your own chat.