Thalassemia in details, pathophysiology, features, investigation and treatment
Reading File
Finding Sources
Finding Sources
Searching PubMed
thalassemia treatment management
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
I have all the information needed for a comprehensive answer. Let me now compile the full detailed response.
Thalassemia - A Comprehensive Review
Definition & Overview
Thalassemia is a group of inherited autosomal codominant disorders caused by germline mutations that reduce the synthesis of either α-globin or β-globin chains. The resulting imbalance in globin chain synthesis leads to anemia through two mechanisms: decreased red cell production (ineffective erythropoiesis) and decreased red cell lifespan (hemolysis). The word thalassemia derives from the Greek thalassa ("sea"), reflecting its original description in Mediterranean populations.
- Robbins, Cotran & Kumar Pathologic Basis of Disease notes that thalassemia is "among the most common inherited disorders of humans."
- Globally, ~1-5% of the world's population carries a thalassemia mutation; approximately 40,000 β-thalassemia patients are born yearly.
- Endemic regions include the Mediterranean basin, Middle East, tropical Africa, the Indian subcontinent, and Southeast Asia - all regions where malaria is or was endemic (heterozygotes are protected from severe P. falciparum malaria).
Genetics & Molecular Basis
Globin Gene Loci
| Gene | Chromosome | Copies |
|---|---|---|
| α-globin (HBA1/HBA2) | Chromosome 16 | 4 (2 per haplotype) |
| β-globin (HBB) | Chromosome 11 | 2 (1 per haplotype) |
Adult hemoglobin A (HbA) = α₂β₂ tetramer.
β-Thalassemia Mutations
Over 100 causative mutations are known, mostly point mutations. They fall into two functional classes:
- β⁰ mutations: Complete absence of β-globin synthesis (nonsense mutations, frameshifts, splice junction defects)
- β⁺ mutations: Reduced but detectable β-globin synthesis (promoter mutations, mild splice site mutations, poly-A signal mutations)
Specific mutation categories (Harrison's 22E):
- Promoter element mutations → mild β⁺ thalassemia
- Exon-intron splice junction mutations → β⁰ and β⁺
- Alternative splice sites introduced into introns/exons → usually β⁺
- 3' polyadenylation signal mutations → mild/silent β⁺
- Initiation codon mutations → β⁰
- Nonsense mutations / frameshift mutations → β⁰ with truncated unstable mRNA
Rare causes include mutations in transcription regulators (SUPT5H, TFIIH) or erythroid factors (GATA1).
α-Thalassemia Mutations
Unlike β-thalassemia, α-thalassemia is caused mainly by gene deletions (not point mutations). Severity is proportional to the number of α-globin genes deleted out of four.
Classification
β-Thalassemia
| Syndrome | Genotype | Clinical Features |
|---|---|---|
| β-Thalassemia major (Cooley's anemia) | Homozygous β⁰/β⁰ or β⁰/β⁺ | Severe anemia; transfusion-dependent |
| β-Thalassemia intermedia | Variable β⁺/β⁺ or β⁻/β⁺ | Moderately severe; not always transfusion-dependent |
| β-Thalassemia minor (trait) | Heterozygous β⁰/normal or β⁺/normal | Mild microcytic anemia; asymptomatic |
| Silent carrier | Very mild β⁺/normal | Normal CBC; carrier state |
Modern classification (Harrison's 22E): Transfusion-dependent (TDT) vs. Non-transfusion-dependent thalassemia (NTDT).
α-Thalassemia
| Syndrome | Genes deleted | Clinical Features |
|---|---|---|
| Silent carrier | −α/αα (1 gene deleted) | Normal; no anemia |
| α-Thalassemia trait | −α/−α or − −/αα (2 genes deleted) | Mild microcytic anemia |
| HbH disease | − −/−α (3 genes deleted) | Moderate hemolytic anemia (Hgb 8-9 g/dL); Hgb H on electrophoresis |
| Hb Bart's hydrops fetalis | − −/− − (all 4 deleted) | Lethal in utero; Hb Bart's (γ₄); severe hydrops |
Pathophysiology
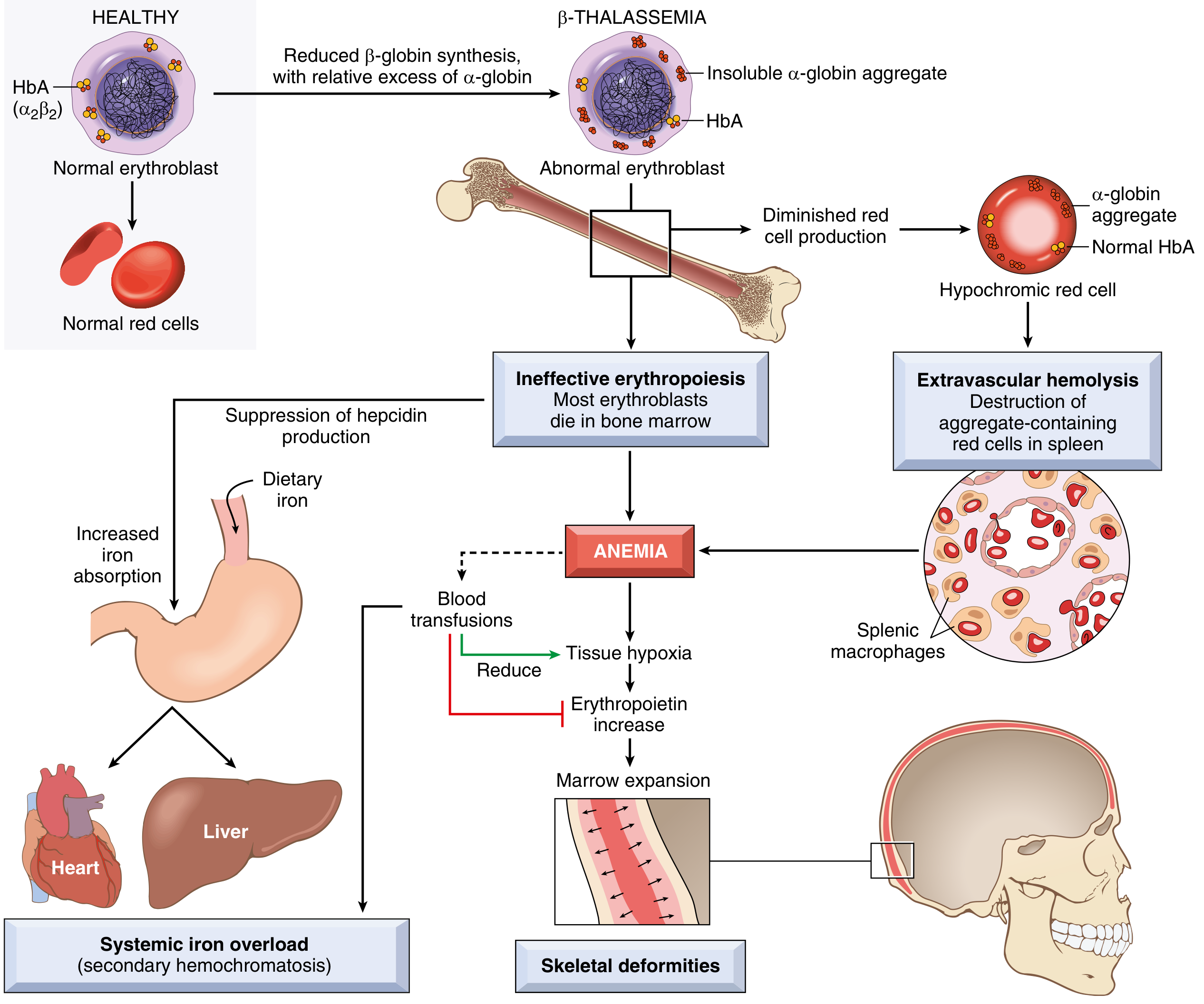
Fig 10.5 - Pathophysiology of β-thalassemia major (Robbins & Kumar Basic Pathology)
β-Thalassemia Pathophysiology
The deficit in β-globin chain synthesis allows α-globin chains to accumulate in excess. Unpaired α-chains are unstable, cannot form stable tetramers, and precipitate within developing erythroblasts, causing:
- Membrane lipid oxidation and damage of erythroid precursors
- Ineffective erythropoiesis (intramedullary destruction of erythroid precursors) - the dominant mechanism of anemia
- Hemolysis - reduced deformability and phosphatidylserine exposure cause extra- and intravascular hemolysis of those erythrocytes that do enter circulation
Cascade of consequences in poorly treated β-thalassemia major:
- Severe anemia → massive compensatory erythroid hyperplasia
- Bone marrow expansion → bony deformities (frontal bossing, "crew-cut" skull on X-ray, maxillary hypertrophy giving "chipmunk facies")
- Hepatosplenomegaly (extramedullary hematopoiesis + RBC trapping)
- Iron accumulation in liver, heart, and endocrine organs (from both transfusions and increased GI absorption driven by ineffective erythropoiesis suppressing hepcidin)
- Pulmonary hypertension and thromboembolic disease
α-Thalassemia Pathophysiology
Loss of α-globin genes leads to excess β-globin chains (in adults) or γ-globin chains (in fetuses/neonates):
- Excess β-chains form β₄ tetramers = Hemoglobin H (HbH)
- Excess γ-chains form γ₄ tetramers = Hemoglobin Bart's (Hb Bart)
Importantly, HbH and Hb Bart cause less membrane damage than free α-chains (because they are more stable tetramers), so ineffective erythropoiesis is less pronounced in α-thalassemia. However, both HbH and Hb Bart have abnormally high oxygen affinity - they cannot release O₂ to tissues, rendering them functionally useless.
With all 4 α-genes deleted: the fetus is entirely dependent on Hb Bart's; severe tissue hypoxia causes massive hydrops fetalis - lethal in utero or very shortly after birth.
Clinical Features
β-Thalassemia Major (Cooley's Anemia)
Usually presents in the first 1-2 years of life when γ→β switch is complete:
Hematological:
- Severe microcytic hypochromic anemia (Hgb often <5 g/dL without transfusion)
- Marked anisopoikilocytosis, target cells, teardrop cells, nucleated RBCs
- Elevated reticulocytes (though erythropoiesis is ineffective overall)
Systemic:
- Failure to thrive, pallor, jaundice
- Massive hepatosplenomegaly (extramedullary hematopoiesis)
- Skeletal deformities: frontal bossing, "chipmunk facies" (maxillary overgrowth), crew-cut skull X-ray appearance, osteoporosis
- Growth retardation and delayed puberty (endocrine iron deposition)
- Gallstones (pigment stones from chronic hemolysis)
Complications of iron overload (in transfused patients):
- Cardiac failure / arrhythmias (most common cause of death)
- Hepatic cirrhosis
- Diabetes mellitus (pancreatic iron)
- Hypogonadism, hypothyroidism, hypoparathyroidism, adrenal insufficiency
- Skin bronzing
β-Thalassemia Intermedia
- Symptomatic anemia (Hgb 7-10 g/dL) but may not require regular transfusions
- Milder skeletal changes; splenomegaly present
- Iron overload still occurs from increased GI absorption
- Normal or late puberty; usually fertile
β-Thalassemia Minor (Trait)
- Usually asymptomatic
- Mild microcytic hypochromic anemia (Hgb rarely < 10 g/dL)
- Elevated RBC count (microcytosis without significant anemia)
- Clinically important only for genetic counseling
HbH Disease (3-gene α-thalassemia)
- Moderate hemolytic anemia (Hgb ~8-9 g/dL)
- Splenomegaly
- Hemolytic crises with infections or oxidant drugs
- Does not usually require regular transfusion
- Rarely transfusion-dependent
Hb Bart's Hydrops Fetalis (4-gene α-thalassemia)
- Stillbirth or death shortly after birth
- Massive hydrops, severe anemia
- Associated with maternal complications (pre-eclampsia, difficult delivery)
Investigations
1. Complete Blood Count (CBC)
- Microcytic hypochromic anemia: low MCV (often <70 fL), low MCH
- Elevated RBC count (in thalassemia trait - distinguishes from iron deficiency)
- Mentzer Index: MCV/RBC count - <13 suggests thalassemia, >13 suggests iron deficiency
2. Peripheral Blood Smear
- Target cells, teardrops, anisopoikilocytosis
- Nucleated RBCs in severe disease
- Basophilic stippling
- HbH inclusions (Heinz bodies-like) in HbH disease (with brilliant cresyl blue stain)
3. Hemoglobin Electrophoresis / HPLC (High-Performance Liquid Chromatography)
- β-thalassemia minor: ↑ HbA₂ (>3.5%, often 4-7%) ± mild ↑ HbF - diagnostic hallmark
- β-thalassemia major: absent or markedly reduced HbA; predominantly HbF (may be >90%)
- HbH disease: HbH band (β₄) on electrophoresis
- Silent α-carrier: usually normal electrophoresis
- β-thalassemia - after recognizing microcytic hypochromic indices and excluding iron deficiency, elevated HbA₂ on HPLC confirms diagnosis
4. Iron Studies
- Serum ferritin, serum iron, TIBC, transferrin saturation
- Important to exclude iron deficiency (which can mask elevated HbA₂) and to monitor iron overload in transfused patients
- Chelation indicated when ferritin consistently >1000 ng/mL
5. Molecular / DNA Analysis
- Definitive diagnosis especially for α-thalassemia (requires gene copy number analysis - multiplex ligation-dependent probe amplification [MLPA] or gap-PCR)
- Identifies specific mutations for genetic counseling and prenatal diagnosis
-
500 unique thalassemia-causing mutations catalogued
6. Imaging
- Skull X-ray: "hair-on-end" or "crew-cut" appearance (marrow expansion through outer table)
- Skeletal survey: generalized osteoporosis, widened medullary cavities
- *MRI (T2)**: gold standard for quantifying cardiac and liver iron overload - critical for managing chelation
- Echocardiography: assess cardiac function in iron-loaded patients
- Ultrasound abdomen: hepatosplenomegaly, gallstones
7. Bone Marrow
- Erythroid hyperplasia
- Usually not required for diagnosis but shows ineffective erythropoiesis
8. Prenatal Diagnosis
- Chorionic villus sampling (CVS) at 10-12 weeks or amniocentesis at 15-18 weeks for DNA analysis
- Cell-free fetal DNA from maternal blood (increasingly available)
- Preimplantation genetic diagnosis (IVF)
Treatment
β-Thalassemia Trait / α-Thalassemia Trait
- No specific treatment required
- Genetic counseling for both partners
- Folic acid supplementation in pregnancy
- Treat concurrent iron deficiency if present (monitor ferritin and transferrin saturation)
- Avoid unnecessary iron supplementation
HbH Disease
- Folic acid 2-5 mg/day
- Avoid oxidant drugs (dapsone, primaquine, sulfonamides)
- Blood transfusions for acute hemolytic crises (with infections)
- Regular monitoring of Hgb and iron stores
β-Thalassemia Major / Transfusion-Dependent Thalassemia (TDT)
1. Regular Red Cell Transfusion
- Goal: maintain pre-transfusion Hgb >9-10 g/dL (Goldman-Cecil: 9-10.5 g/dL)
- Every 2-5 weeks (usually every 3-4 weeks)
- Use leukoreduced, phenotypically matched packed red cells (to minimize alloimmunization and transfusion reactions)
- Suppresses ineffective erythropoiesis and prevents skeletal deformities
- Decision to start lifelong transfusion: based on definitive diagnosis, molecular defects, severity of anemia on repeated measurement, clinical criteria (failure to thrive, bone changes)
2. Iron Chelation Therapy
Iron overload from repeated transfusions is the leading cause of morbidity/mortality. Chelation is indicated when ferritin consistently >1000 ng/mL (after ~20 units of pRBCs):
| Agent | Route | Dose | Notes |
|---|---|---|---|
| Deferoxamine (DFO, Desferal) | SC/IV infusion 8-12 hours overnight | 40 mg/kg | Gold standard; not oral; poor adherence |
| Deferasirox (Exjade/Jadenu) | Oral daily | Exjade: 20-40 mg/kg/d; Jadenu: 7-21 mg/kg/d | GI side effects; renal monitoring needed |
| Deferiprone (L1) | Oral 3x/day | 75-100 mg/kg/d | Agranulocytosis risk; effective for cardiac iron; often combined with DFO |
- Target ferritin <1000 ng/mL; MRI T2* >20 ms (liver), >20 ms (heart) indicates adequate chelation
- Luspatercept (Reblozyl) 1 mg/kg SC every 3 weeks - approved for TDT to reduce transfusion requirements; acts as erythroid maturation agent (TGF-β trap); median time to response 12-24 days
3. Splenectomy
- Considered when annual blood consumption increases progressively (>2 units/month or increasing transfusion requirements)
- Reduces hypersplenism and transfusion requirements
- Risks: post-splenectomy sepsis (vaccinate against pneumococcus, H. influenzae, N. meningitidis at least 2 weeks before surgery), thromboembolic events, pulmonary hypertension
- Avoid in children <5-6 years due to high infection risk
- Lifelong prophylactic penicillin post-splenectomy
4. Folic Acid
- 1-5 mg/day to support erythropoiesis
5. Hematopoietic Stem Cell Transplantation (HSCT)
- Only proven curative therapy
- Best results in young patients (<14 years), Pesaro Class I-II (no hepatomegaly, regular chelation, no portal fibrosis)
- Requires HLA-matched sibling donor (best outcome); matched unrelated donor possible
- Overall cure rate ~80-90% in low-risk patients with matched sibling
- Risks: graft-versus-host disease, graft failure, conditioning regimen toxicity
6. Gene Therapy (Emerging/Approved)
- Betibeglogene autotemcel (Zynteglo): lentiviral vector delivering functional β-globin gene; FDA-approved; can achieve transfusion independence in many TDT patients
- Exagamglogene autotemcel (Casgevy): CRISPR/Cas9 gene editing to reactivate fetal hemoglobin (HbF) by disrupting BCL11A erythroid enhancer; recently approved
- Very costly; curative potential but access limited in low-resource settings
7. Hydroxyurea
- Increases HbF production; more useful in β-thalassemia intermedia/NTDT
- Reduces transfusion requirements in some patients
Monitoring in TDT
- Regular Hgb, ferritin, LFTs, renal function
- Annual/biannual MRI T2* (liver and cardiac iron)
- Echocardiography annually
- Endocrine assessment (glucose, thyroid, parathyroid, pubertal development)
- Ophthalmology and audiology (deferoxamine toxicity screening)
Summary Table: Key Points
| Feature | β-Thal Major | β-Thal Minor | HbH Disease | Hb Bart's |
|---|---|---|---|---|
| Genes affected | Both β-globin | One β-globin | 3 α-globin | All 4 α-globin |
| HbA₂ | Low (HbF dominates) | ↑ (4-7%) | Normal | Absent |
| HbF | Very high | Mildly ↑ | Normal/mildly ↑ | 0 |
| Anemia severity | Severe | Mild | Moderate | Lethal |
| Transfusion-dependent | Yes | No | Rarely | N/A |
| Treatment | Transfusion + chelation ± HSCT | None (counseling) | Folic acid; crisis management | Intrauterine transfusion (experimental) |
Sources
- Harrison's Principles of Internal Medicine, 22nd Ed. (2025), Chapter 103 - Hemoglobinopathies
- Robbins, Cotran & Kumar Pathologic Basis of Disease - Chapter 14
- Robbins & Kumar Basic Pathology - Chapter 10
- Goldman-Cecil Medicine - Thalassemia chapter
- Washington Manual of Medical Therapeutics - Hematology section
Nephrotic syndrome in children, pathophysiology diagnosis, features and management
Reading File
Finding Sources
Finding Sources
Searching PubMed
"nephrotic syndrome" children management
Finding Sources
Reading Article
PMID: 40254391
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
I now have rich, comprehensive content from multiple authoritative textbooks. Let me compile the full detailed answer.
Nephrotic Syndrome in Children - Comprehensive Review
Definition
Nephrotic syndrome (NS) is a clinical constellation defined by:
- Proteinuria >40 mg/m²/hour or urinary protein:creatinine ratio >2.0 mg/mg (in children); >3.5 g/24h in adults
- Hypoalbuminemia <2.5 g/dL (serum albumin <3.5 g/dL)
- Generalized edema (anasarca)
- Hyperlipidemia (secondary)
Epidemiology
- Incidence: ~2-7 per 100,000 children per year; prevalence ~16 per 100,000
- Minimal change nephrotic syndrome (MCNS) accounts for ~90% of cases in children <10 years of age and 70-80% of all pediatric NS
- Peak incidence at 2-5 years of age (preschool children); median age 2.5 years
- Male:Female ratio approximately 2:1 in childhood (equalizes in adolescence)
- Steroid-sensitive NS (SSNS): ~80-90% of children; steroid-resistant NS (SRNS): 15-20%
Classification
By Age of Onset
| Age | Category | Notes |
|---|---|---|
| 0-3 months | Congenital NS | Usually genetic; non-responsive to immunosuppression |
| 3-12 months | Infantile NS | Often genetic; consider Finnish-type (NPHS1 mutation) |
| 1-8 years | Childhood NS | Predominantly MCNS; steroid-sensitive |
| >8 years | Older child/adolescent | More FSGS, secondary causes |
By Etiology
- Primary (Idiopathic): MCNS, FSGS, Membranous nephropathy (MN), Membranoproliferative GN (MPGN)
- Secondary: Systemic disease (SLE, HSP/IgAV, amyloidosis, diabetes, HBV, HCV, malaria, HIV, syphilis)
- Genetic/Hereditary: Mutations in ~30 genes (NPHS1, NPHS2, WT1, PLCE1, LAMB2, CD2AP, TRPC6, ACTN4, etc.)
- Drug-induced: NSAIDs, captopril, lithium, penicillamine, heroin
By Histopathology (Primary NS)
| Histology | Frequency in Children | Steroid Response |
|---|---|---|
| Minimal Change Disease (MCD) | 70-90% | ~90% response |
| Focal Segmental Glomerulosclerosis (FSGS) | Most common in SRNS | <25% response |
| Membranous nephropathy (MN) | Rare in children | Variable |
| Membranoproliferative GN (MPGN) | Uncommon | Variable |
By Steroid Response (Clinically Most Useful)
- Steroid-Sensitive NS (SSNS): remission within 4-8 weeks of full-dose prednisone
- Steroid-Resistant NS (SRNS): failure to achieve remission after 6-8 weeks of full-dose daily prednisone
- Frequently Relapsing NS (FRNS): ≥2 relapses within 6 months of initial response, OR ≥4 relapses within any 12-month period
- Steroid-Dependent NS (SDNS): relapses during steroid tapering or within 2 weeks of stopping steroids on two consecutive occasions
Pathophysiology
The Glomerular Filtration Barrier
The normal filtration barrier has three layers:
- Fenestrated glomerular endothelium - size and charge barrier
- Glomerular basement membrane (GBM) - rich in negatively charged heparan sulfate proteoglycans
- Podocytes (visceral epithelial cells) with foot processes connected by the slit diaphragm (nephrin, podocin, CD2AP proteins)
Increased permeability from structural or physicochemical alterations in this barrier allows proteins to escape into the urinary space.
Core Mechanism: Proteinuria → Cascade
The nephrotic cascade is initiated by glomerular protein leak, leading to a chain of consequences:
Glomerular barrier injury (podocyte/slit diaphragm dysfunction)
↓
MASSIVE PROTEINURIA (predominantly albumin)
↓
HYPOALBUMINEMIA (urinary loss > hepatic synthetic capacity + renal catabolism)
↓
↓ Plasma oncotic pressure
↓
Fluid shifts from intravascular → interstitial space (UNDERFILL theory)
↓
Perceived low effective circulating volume
↓
RAAS activation + ↑ ADH + ↑ sympathetic tone → Na⁺ + H₂O retention
↓
EDEMA
Two theories of edema formation:
- Underfill theory (classic, especially in MCD): low albumin → ↓ oncotic pressure → plasma volume contracted → secondary Na retention. RAAS activated, renin and aldosterone raised.
- Overfill theory: primary intrinsic renal Na retention (especially in FSGS and membranous nephropathy) → expanded plasma volume → secondary edema. RAAS suppressed.
In practice, many patients have elements of both. Children with MCD typically manifest the underfill pattern with contracted plasma volume and activated RAAS.
Pathogenesis of MCD (Most Important in Children)
- Light microscopy: normal glomeruli - hence "minimal change"
- Electron microscopy: diffuse effacement (fusion) of podocyte foot processes - pathognomonic
- Immunofluorescence: negative (no immune complex deposits)
- Pathogenic mechanism: autoantibodies against the slit diaphragm protein nephrin are now recognized in many cases (Goldman-Cecil); T-cell dysfunction with circulating permeability factor (historically postulated); angiopoietin-like-4 (ANGPTL4) secreted by podocytes has been shown to mediate proteinuria in glucocorticoid-sensitive NS
Secondary Consequences
| Consequence | Mechanism |
|---|---|
| Hyperlipidemia | ↑ hepatic lipoprotein synthesis (response to ↓ oncotic pressure); ↓ lipid catabolism (loss of lipoprotein lipase); ↑ VLDL, LDL, total cholesterol; ↓ HDL |
| Lipiduria | Lipoproteins leak through glomerulus; appear as oval fat bodies and fatty casts (Maltese cross pattern under polarized light) |
| Hypercoagulability | Loss of antithrombin III, protein C, protein S, plasminogen in urine; ↑ fibrinogen (acute phase reactant); ↑ platelet aggregability → thrombosis risk |
| Susceptibility to infection | Loss of IgG and complement factors (opsonins) in urine; T-cell dysfunction; edematous skin barrier breakdown → especially pneumococcal, staphylococcal peritonitis/sepsis |
| Iron deficiency anemia | Loss of transferrin |
| Vitamin D deficiency | Loss of vitamin D-binding protein |
| Hypothyroidism (subclinical) | Loss of thyroid-binding globulin |
| AKI | Low effective circulating volume → prerenal; or in heavy proteinuria → tubular injury |
Clinical Features
Presenting Symptoms
- Periorbital edema - classically the first sign; worse in the morning; often mistaken for allergy
- Pitting edema - dependent; lower limbs, scrotum/labia
- Ascites - abdominal distension, discomfort
- Pleural effusions - dyspnea (if large)
- Weight gain (fluid)
- Reduced urine output (concentrated, frothy/foamy urine)
- Pallor, lethargy, anorexia
Signs
- Soft pitting edema - periorbitally, legs, scrotum/labia, sacrum
- Ascites (shifting dullness, fluid thrill)
- Blood pressure - usually normal in MCD; hypertension is atypical and should prompt further investigation
- No hematuria (typically) - microscopic hematuria present in ~20% of MCD; macroscopic hematuria is unusual and suggests nephritic component or FSGS/MN
Features Atypical for MCD (Warranting Biopsy Before Steroids)
- Age <1 year at presentation (especially <3 months - congenital NS)
- Macroscopic hematuria
- Persistent hypertension
- Hypocomplementemia (↓C3/C4 - suggests MPGN, lupus, post-infectious)
- Extrarenal features: rash, arthritis, hepatosplenomegaly (secondary causes)
- Impaired renal function (elevated creatinine)
- Positive ANA/ANCA/anti-dsDNA
- Failure to respond to 8 weeks of steroids (SRNS)
- Strong family history (genetic NS)
Investigations
1. Urine Tests
- Urine dipstick: ≥3+ protein (+++); may show lipiduria
- Spot urine protein:creatinine ratio (PCR): >2 mg/mg (200 mg/mmol) confirms nephrotic-range proteinuria
- 24-hour urine protein: >40 mg/m²/h or >3.5 g/day; largely replaced by spot PCR in children
- Urinalysis with microscopy: oval fat bodies, fatty casts, Maltese cross birefringence; RBC casts absent in pure NS (if present, suggests nephritic component)
- Urine sodium: low in underfill (hypovolemic) state
2. Blood Tests
| Test | Expected Finding | Significance |
|---|---|---|
| Serum albumin | <2.5 g/dL (often <1.5 g/dL in severe) | Confirms hypoalbuminemia |
| Total protein | Low | |
| Lipid profile | ↑↑ Total cholesterol, LDL, triglycerides; ↓ HDL | Hyperlipidemia |
| Renal function (urea, creatinine, eGFR) | Usually normal in MCD | ↑ in SRNS/FSGS or hypovolemia |
| Serum electrolytes | Na⁺ may be low (dilutional); K⁺ variable | Monitor during diuretic therapy |
| FBC | Normocytic anemia (transferrin loss); ↑ platelets | |
| Complement (C3, C4) | Normal in MCD/FSGS; ↓ in MPGN, lupus, post-infectious | Differentiates causes |
| ANA, anti-dsDNA | For lupus nephritis | Especially girls >8 years |
| ANCA | For pauci-immune GN | |
| Anti-PLA2R | For membranous nephropathy | |
| HBsAg, HCV, HIV | Secondary causes | |
| Blood culture | For sepsis during relapse | |
| PT/APTT, fibrinogen, antithrombin III | Hypercoagulable state assessment | |
| Thyroid function, Vitamin D | Often depleted in NS | |
| Genetic testing | SRNS or age <1 year; congenital NS | Next-generation sequencing |
3. Imaging
- Renal ultrasound: enlarged, echogenic kidneys in NS (non-specific); rules out obstruction, cysts; assesses for thrombosis (Doppler if renal vein thrombosis suspected)
- Chest X-ray: if dyspneic - pleural effusions
- Abdominal X-ray/ultrasound: ascites
4. Kidney Biopsy
Indications in children:
- Age <1 year (congenital/infantile NS)
- SRNS (failure after 8 weeks of full-dose steroids)
- Atypical features (macroscopic hematuria, persistent hypertension, hypocomplementemia, extrarenal disease)
- Suspicion of secondary cause
- Frequently relapsing NS being considered for second-line therapy
Not required for typical childhood presentation (age 1-8 years, no atypical features) - treat empirically with steroids.
Biopsy findings:
- MCD: LM normal; EM - diffuse podocyte foot process effacement; IF - negative
- FSGS: LM - focal (<50% glomeruli) and segmental sclerosis/hyalinosis; EM - foot process effacement; IF - IgM/C3 in sclerotic segments
- Membranous: LM - GBM thickening, subepithelial "spikes"; EM - subepithelial immune deposits; IF - granular IgG/C3 (PLA2R staining positive in primary MN)
- MPGN: LM - mesangial proliferation + GBM "tram-track" splitting; IF - C3 ± Ig deposits
Management
General Principles
- Confirm diagnosis (urine PCR, serum albumin, lipids)
- Assess for atypical features requiring biopsy
- Educate family on urine dipstick monitoring at home
- Low-salt diet during edema phase
- Restrict fluid if hyponatremic
- Monitor weight daily; input/output chart
1. Corticosteroids - First-Line Treatment
Initial episode (KDIGO 2025 / Standard protocols):
- Prednisolone 60 mg/m²/day (max 60 mg/day) as a single morning dose for 4-6 weeks (daily)
- Then 40 mg/m² on alternate days for 4-6 weeks, then taper over 4-8 weeks
- Total initial treatment duration: minimum 12-16 weeks (longer courses reduce relapse risk - evidence from RCTs)
- KDIGO 2025 recommends extended initial steroid course
Relapse treatment:
- Prednisolone 60 mg/m²/day until urine protein-free for 3 consecutive days
- Then 40 mg/m² on alternate days for 4 weeks, then stop
Definitions of response:
- Remission: urine protein dipstick trace or negative (or PCR <0.2) for 3 consecutive days
- Relapse: urine protein ≥2+ (or PCR >2) for 3 consecutive days after remission
- SSNS: complete remission within 8 weeks
- SRNS: no remission after 8 weeks of daily high-dose steroids
2. Management of Edema
- Mild edema: salt restriction alone (no added salt diet, ~1-2 g/day in children)
- Moderate-severe edema: Furosemide 1-2 mg/kg/dose oral or IV (loop diuretic)
- Furosemide + spironolactone combination for resistant edema
- IV albumin (20-25%, 0.5-1 g/kg over 2-4 hours) + IV furosemide for severe refractory edema with signs of intravascular volume depletion/hypovolemia - use cautiously (risk of pulmonary edema)
- Avoid vigorous diuresis in hypovolemic patients (risk of AKI, thrombosis)
3. Steroid-Sparing Agents (FRNS/SDNS)
| Agent | Dose | Notes |
|---|---|---|
| Levamisole | 2.5 mg/kg on alternate days | Cheap; immunomodulator; used in LMIC; ↑ relapse risk when stopped |
| Oral cyclophosphamide | 2-3 mg/kg/day for 8-12 weeks | Alkylating agent; sustained remission in 50%; cumulative gonadotoxic dose limit |
| Mycophenolate mofetil (MMF) | 600-1200 mg/m²/day in 2 divided doses | Well tolerated; commonly used; less effective than cyclophosphamide |
| Calcineurin inhibitors (CNIs): Cyclosporine or Tacrolimus | Cyclosporine: 4-5 mg/kg/day; Tacrolimus: 0.05-0.15 mg/kg/day | Effective but renal toxicity with long use; high relapse rate on stopping |
| Rituximab (anti-CD20) | 375 mg/m² IV × 1-4 doses | B-cell depletion; effective in FRNS/SDNS and some SRNS; increasingly used |
| Voclosporin / Sparsentan | Newer agents | Emerging; mainly studied in adults/FSGS |
4. Steroid-Resistant NS (SRNS) Management
- Kidney biopsy mandatory (exclude FSGS, MN)
- Genetic testing (NPHS1, NPHS2, WT1 mutations especially in young children)
- CNIs (cyclosporine or tacrolimus) - first-line for SRNS after biopsy
- Pulse IV methylprednisolone (30 mg/kg/dose, max 1 g) may be tried
- Rituximab in some cases
- High-dose prednisone for 4-16 weeks
- Genetic SRNS: no response to immunosuppression; focus on supportive care
5. Congenital NS Management
- Immunosuppression is not recommended (non-responsive)
- Conservative management: Na and fluid restriction, intermittent IV albumin + furosemide
- Hypercaloric diet for growth
- Thyroid hormone replacement (TBG loss)
- Monitor/treat thrombotic episodes
- Aggressive management of infections
- Bilateral nephrectomy + renal replacement therapy (dialysis → transplant) as definitive management
6. Management of Complications
| Complication | Management |
|---|---|
| Infection (spontaneous bacterial peritonitis - SBP, cellulitis, pneumococcal sepsis) | Pneumococcal vaccine (ideally during remission); prompt broad-spectrum antibiotics; avoid live vaccines during immunosuppression |
| Thromboembolism (DVT, pulmonary embolism, renal vein thrombosis) | Prophylactic anticoagulation if albumin <2 g/dL + heavy proteinuria; therapeutic anticoagulation (LMWH → warfarin) for confirmed thrombosis; maintain adequate hydration; avoid prolonged immobility |
| AKI / Hypovolemia | IV fluid resuscitation (NS bolus); IV albumin if hypoalbuminemia-driven; withhold diuretics; monitor renal function |
| Hyperlipidemia | Usually resolves with remission; statins if persistent (used cautiously in children) |
| Hypertension | ACE inhibitors (enalapril, ramipril) or ARBs - also antiproteinuric; amlodipine |
| Vitamin D deficiency | Cholecalciferol supplementation |
| Steroid side effects | Cushingoid features, growth retardation, hypertension, glucose intolerance, cataracts, osteoporosis, behavioral changes → use minimum effective dose; alternate-day dosing |
7. ACE Inhibitors/ARBs
- Not typically used for induction of remission in SSNS
- Useful for: SRNS, FSGS, persistent proteinuria, hypertension, renal protection
- Reduce proteinuria and slow progression of CKD
- Monitor potassium and creatinine; avoid if AKI or hypovolemia
Prognosis
| Type | Prognosis |
|---|---|
| SSNS (MCD) | Excellent; ~85% achieve long-lasting remission; ~50% relapse at least once; <5% progress to CKD |
| FRNS/SDNS | Good with steroid-sparing agents; quality of life can be significantly impacted |
| SRNS (FSGS) | Guarded; 50-80% progress to ESRD within 5-10 years if no remission |
| Congenital NS (Finnish type) | Severe; most require transplantation by age 2-3 years |
| Genetic SRNS (NPHS2, WT1) | Variable; transplantation is curative (no recurrence if genetic cause) |
| Primary FSGS | Recurs in transplant in 30-40% of cases |
Key Summary Points
- In children 1-8 years, MCD is the presumptive diagnosis - empirical steroid therapy without biopsy is standard
- The tetrad = massive proteinuria + hypoalbuminemia + edema + hyperlipidemia
- First-line = prednisolone 60 mg/m²/day × 4-6 weeks, then alternate-day taper
- ~90% of children with MCD are steroid-sensitive
- Biopsy is indicated for atypical features, SRNS, or before second-line therapy
- Complications to watch: spontaneous bacterial peritonitis, thromboembolism, AKI from hypovolemia, steroid toxicity
- KDIGO 2025 is the current guideline for management of nephrotic syndrome in children (PMID: 40254391)
Sources
- Campbell-Walsh-Wein Urology - Pediatric Glomerular Disease chapter
- Robbins, Cotran & Kumar Pathologic Basis of Disease - Nephrotic Syndrome chapter
- NKF Primer on Kidney Diseases, 8th Ed. - Nephrotic Syndrome
- Goldman-Cecil Medicine - Idiopathic Nephrotic Syndrome
- Brenner and Rector's The Kidney - Sodium/fluid retention in NS
- Symptom to Diagnosis, 4th Ed. - Evidence-Based Diagnosis
- KDIGO 2025 Clinical Practice Guideline for Management of Nephrotic Syndrome in Children (PMID: 40254391) - current authoritative guideline
This is a shared conversation. Sign in to Orris to start your own chat.