Micheal aplasia and mondini deformity. MS ent 20 marks University exam pattern.
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Reading File
Loading Image
I now have comprehensive information to write a detailed MS ENT 20-mark university exam answer on both topics. Let me compose the full answer.
Michel Aplasia and Mondini Deformity
MS ENT - 20 Marks University Exam Answer
INTRODUCTION - Inner Ear Malformations
Inner ear malformations account for approximately 20% of congenital sensorineural hearing loss (SNHL). They are classified based on the timing of embryological developmental arrest. The otic cyst (vesicle) develops three buds between weeks 4-5 that form the cochlea, semicircular canals (SCCs), and vestibular aqueduct (VA). The five classical types in order of severity are:
| Malformation | Incidence |
|---|---|
| Incomplete partition (Mondini dysplasia) | 55% |
| Common cavity | 26% |
| Cochlear hypoplasia | 15% |
| Cochlear aplasia | 3% |
| Complete labyrinthine aplasia (Michel aplasia) | 1% |
(Cummings Otolaryngology, Table 194.1)
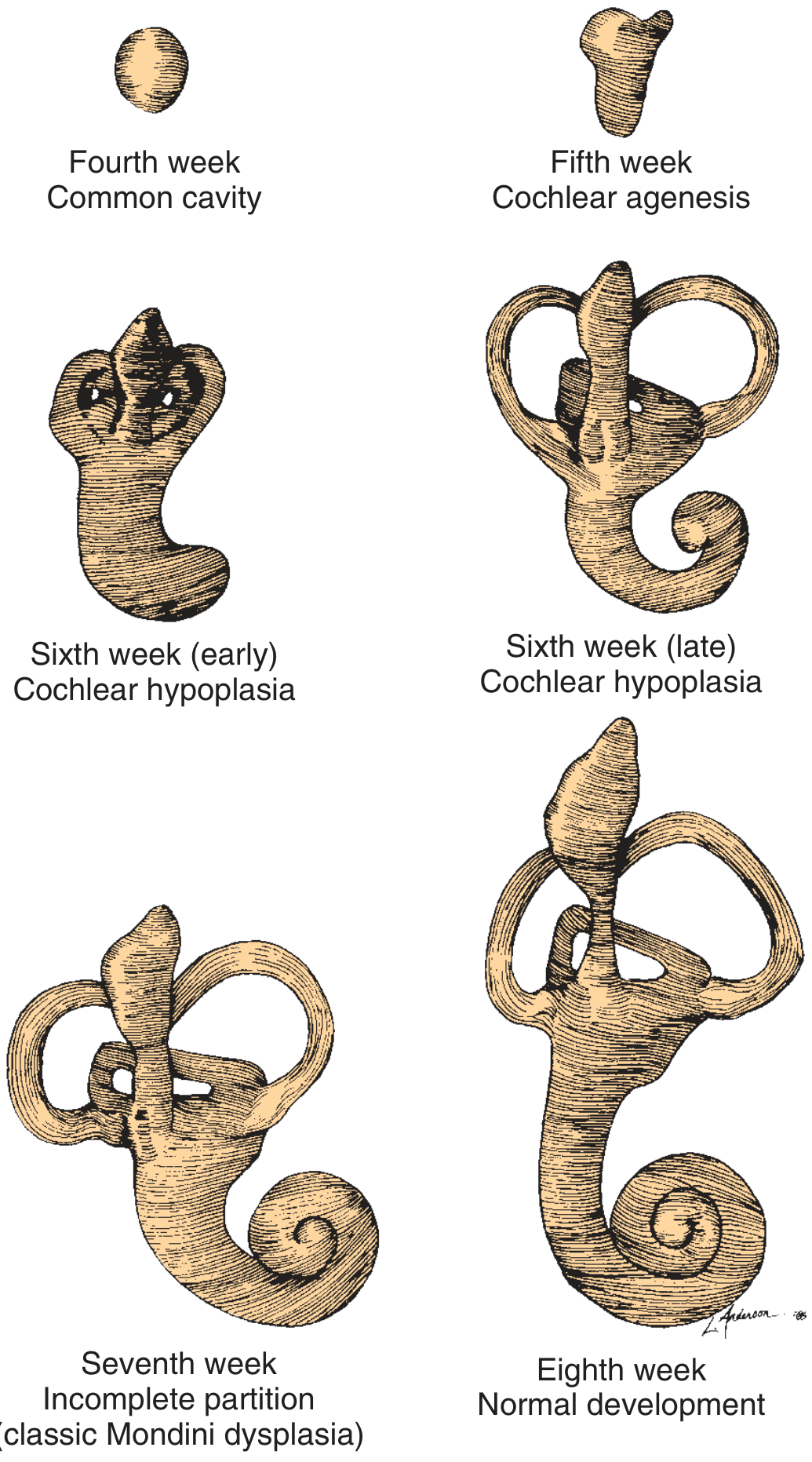
PART I: MICHEL APLASIA (Complete Labyrinthine Aplasia)
Definition
Michel aplasia is the most severe form of inner ear malformation, characterized by complete absence of both the membranous and osseous labyrinth.
Historical Note
First described by Michel in 1863. The complete absence of the otic capsule means these ears are uniformly deaf (anacusis).
Embryology / Pathogenesis
- Results from a developmental arrest before the formation of the otic vesicle - i.e., before the end of the 3rd gestational week (some sources say before week 4)
- The otic placode fails to invaginate, so no otic vesicle forms
- The entire bony and membranous labyrinth is absent
- The external and middle ear may be unaffected (they develop from separate branchial arch structures)
Pathology
- Complete agenesis of the petrous portion of the temporal bone
- No cochlea, no vestibule, no semicircular canals, no internal auditory canal (IAC)
- The otic capsule is entirely absent
- External ear canal and middle ear may be present normally
Radiology (HRCT temporal bone - key investigation)
- Complete absence of the otic capsule on axial and coronal HRCT
- Ear canal and middle ear present but inner ear structures absent
- MRI confirms absence of 8th nerve in IAC (aplasia of vestibulocochlear nerve often coexists)
- Must be differentiated from labyrinthine ossification - in ossification, the otic capsule is dense and of normal dimensions; in Michel aplasia, the otic capsule bone is absent
Audiological Features
- Profound bilateral SNHL (anacusis / total deafness) from birth
- No response on ABR (Auditory Brainstem Response)
- No otoacoustic emissions
Genetics
- Autosomal dominant (AD) inheritance has been observed
- Autosomal recessive inheritance also reported
- Inherited complete labyrinthine aplasia has been described
Management
- Conventional hearing aids - no benefit (no cochlea or auditory nerve)
- Cochlear implant (CI) - CONTRAINDICATED (no cochlea or nerve)
- Auditory Brainstem Implant (ABI) - may be attempted if cochlear nerve absent
- Vibrotactile devices - have proven beneficial in some patients (Lee's)
- Special education and rehabilitation for deaf-blind communication
- Genetic counseling for family
PART II: MONDINI DEFORMITY (Incomplete Partition)
Definition and Historical Note
Mondini deformity (incomplete partition / Mondini dysplasia) is the most common cochlear malformation, accounting for >50% of all cochlear anomalies.
Carlo Mondini (1791) first described the deformity in a deaf 8-year-old boy who died after being struck by a wagon. The cochlea showed only 1.5 turns with a hollow apical cavity, an enlarged vestibule, and an enlarged vestibular aqueduct.
Embryology / Pathogenesis
- Developmental arrest at the 7th week of gestation
- At this stage the cochlea is forming its final coils; arrest produces only 1.5 turns instead of the normal 2.5-2.75 turns
- The interscalar septum fails to develop in the apical turn
- Results in a scala communis - the middle and apical turns merge into a single cystic cavity
Pathology
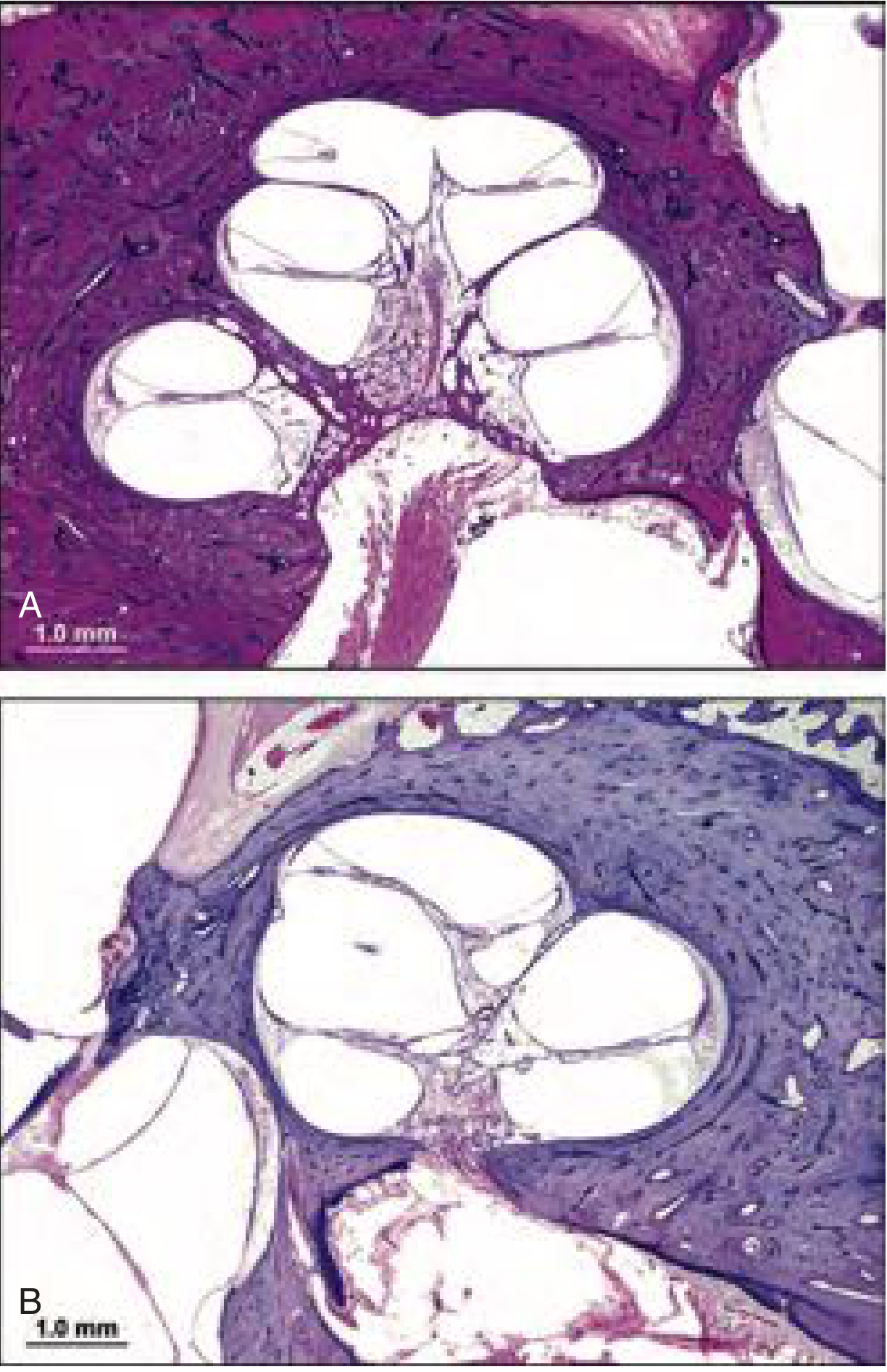
The classic triad (Mondini's original description):
- Cochlea with only 1.5 turns (normally 2.5-2.75 turns)
- Absent interscalar septum in the apical portion - forming an "apical scala communis"
- Enlarged vestibular aqueduct (EVA) and endolymphatic duct/sac
Additional features:
- Enlarged vestibule
- Deficient or absent modiolus (bony core of cochlea)
- Variable organ of Corti development
- Variable spiral ganglion neuron population
- Perilymphatic-endolymphatic communication abnormality due to defect in the cribriform area of the lateral IAC wall - leads to perilymphatic fistula risk
Histopathology (Photomicrograph)
Sennaroglu Classification of Incomplete Partition (3 subtypes)
| Type | Features |
|---|---|
| Type I (IP-I) | Complete absence of modiolus and interscalar septa; cochlea resembles a cyst; usually accompanied by cystic vestibule |
| Type II (IP-II / Classic Mondini) | Normal basal turn + cystic apex; deficient modiolus; enlarged vestibular aqueduct; most common |
| Type III (IP-III) | Deficient modiolus with partial interscalar septation at the cochlea's periphery |
Radiological Features (HRCT Temporal Bone)
- Cochlea smaller than normal: normally 8-10 mm vertically; in Mondini typically 5-6 mm vertically
- 1.5 cochlear turns (instead of 2.5-2.75)
- Absent interscalar septum (more evident on CT than MRI)
- Absent or deficient modiolus
- Enlarged vestibular aqueduct (midpoint diameter > 1.5 mm)
- Enlarged vestibule
- Semicircular canal deformities in ~20% of cases
- HRCT is the imaging of choice; MRI (T2 fast spin echo) shows internal detail
Audiological Features
- Variable hearing loss - from normal to profound SNHL
- Mean hearing threshold ~75 dB (three-tone average) in a series of 41 ears (Cummings)
- Fluctuating hearing loss - a hallmark, due to CSF-perilymph pressure changes through the fistula
- May be unilateral
- "Gusher" phenomenon - perilymph gusher on stapedotomy (due to widened IAC and abnormal communication with CSF)
Genetics and Associations
- Autosomal dominant inheritance described
- Autosomal recessive Mondini dysplasia also reported
- DFN3 locus deletions - associated with familial Mondini dysplasia (X-linked)
- Described in multiple syndromes:
- Pendred syndrome (goiter + SNHL)
- Waardenburg syndrome
- Treacher Collins syndrome
- Wildervanck syndrome (Cervico-oculo-acoustic syndrome)
- CHARGE syndrome (Coloboma, Heart defects, choanal Atresia, Retardation, Genital hypoplasia, Ear anomalies) - Mondini-type deformity + absent SCCs
- Non-genetic: Congenital CMV infection - may account for >40% of deafness of unknown etiology
Complications
- Recurrent meningitis - due to abnormal CSF-perilymph communication via cribriform area of IAC
- Perilymphatic fistula - more common than in normal inner ear
- Perilymph gusher during ear surgery
- Progressive SNHL
Management
1. Audiological Rehabilitation:
- Early hearing aid fitting (most have residual hearing)
- Early audiological assessment (ABR, OAE, ASSR)
- The presence of neurosensory structures in most cases warrants early rehabilitative intervention with conventional amplification
2. Cochlear Implantation (CI):
- Most patients with Mondini deformity are candidates for CI (presence of cochlear nerve is the key criterion, not cochlear anatomy)
- Requires special surgical considerations:
- Risk of perilymph gusher (profuse CSF flow on cochleostomy) - managed by gentle packing with fascia
- Risk of facial nerve anomalies
- Modified insertion technique may be needed
- Full electrode insertion is usually possible
- Outcomes with CI are generally good
3. Surgical precautions:
- Avoid unnecessary middle ear surgery (stapedotomy risk of perilymph gusher)
- Cover any perilymphatic fistula if suspected
- In case of recurrent meningitis - close fistula/seal IAC
4. Meningitis prophylaxis:
- Pneumococcal and Hib vaccination recommended (as in all inner ear malformation patients)
- Antibiotic prophylaxis for recurrent meningitis may be considered
COMPARISON TABLE: Michel Aplasia vs. Mondini Deformity
| Feature | Michel Aplasia | Mondini Deformity |
|---|---|---|
| Other name | Complete labyrinthine aplasia | Incomplete partition (IP) |
| Incidence among cochlear anomalies | 1% | 55% |
| Gestational arrest | Before 3rd week | 7th week |
| Cochlear structure | Completely absent | 1.5 turns, cystic apex |
| Modiolus | Absent | Absent/deficient |
| Interscalar septum | N/A | Absent in apex |
| Vestibular aqueduct | N/A | Enlarged |
| Middle/external ear | May be normal | Usually normal |
| Audiogram | Anacusis (total deafness) | Variable SNHL (mean 75 dB) |
| Genetics | AD (mostly) | AD, AR, DFN3 locus |
| Key syndromes | - | Pendred, Waardenburg, CHARGE |
| Hearing aid | No benefit | Beneficial in most |
| Cochlear implant | Contraindicated | Possible (with precautions) |
| Key risk | - | Meningitis, perilymph gusher |
| Radiology | Absent otic capsule | 1.5 turns, absent septum, EVA |
ADDITIONAL CONTEXT: Classification of Inner Ear Aplasias
For completeness, the 5 classical aplasias (Cummings/Lee's):
- Michel aplasia - arrest before week 3; complete absence of inner ear; anacusis
- Common cavity - arrest at week 4; cochlea + vestibule form one undifferentiated cystic space
- Cochlear aplasia - arrest at week 5; absent cochlea, vestibule + SCCs present
- Cochlear hypoplasia - arrest at week 6; small cochlear bud (1 turn or less); variable hearing
- Mondini (incomplete partition) - arrest at week 7; 1.5-turn cochlea; cystic apex; most common
The classical membranous labyrinth aplasias (histopathological):
- Scheibe aplasia (cochleosaccular dysplasia) - most common; AR; bony labyrinth normal; associated with Usher, Refsum, Jervell-Lange-Nielsen, congenital rubella
- Alexander aplasia - basal coil organ of Corti affected; high-frequency SNHL; amplification beneficial
Sources:
- Cummings Otolaryngology Head and Neck Surgery, Chapter 194 (Inner Ear Malformations)
- K.J. Lee's Essential Otolaryngology, Chapter on Inner Ear Structural Malformations
This is a shared conversation. Sign in to Orris to start your own chat.