Teach me everything i need to know about Hepatorenal syndrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
"hepatorenal syndrome" AND management
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
I now have rich, comprehensive content from multiple authoritative textbooks. Let me compile the complete guide on Hepatorenal Syndrome.
Hepatorenal Syndrome (HRS): A Complete Guide
1. Definition and Overview
Hepatorenal Syndrome (HRS) is a form of functional renal failure without intrinsic renal pathology that occurs in patients with advanced cirrhosis, acute liver failure, or severe alcoholic hepatitis. The kidneys are histologically normal - renal function may be restored by correcting portal hypertension, liver transplantation (LT), or even by transplanting the same kidneys into a non-cirrhotic recipient.
- Occurs in ~10-30% of hospitalized cirrhotics with ascites
- Annual frequency in cirrhotics with ascites: ~8% (some reports up to 40%)
- Develops in ~30% of cirrhotics admitted with SBP, ~25% with severe alcoholic hepatitis, ~10% requiring serial large-volume paracenteses
(Sleisenger and Fordtran's GI and Liver Disease, Harrison's Principles of Internal Medicine 22E)
2. Pathophysiology
The pathophysiology involves three interacting components that all converge to reduce renal perfusion:
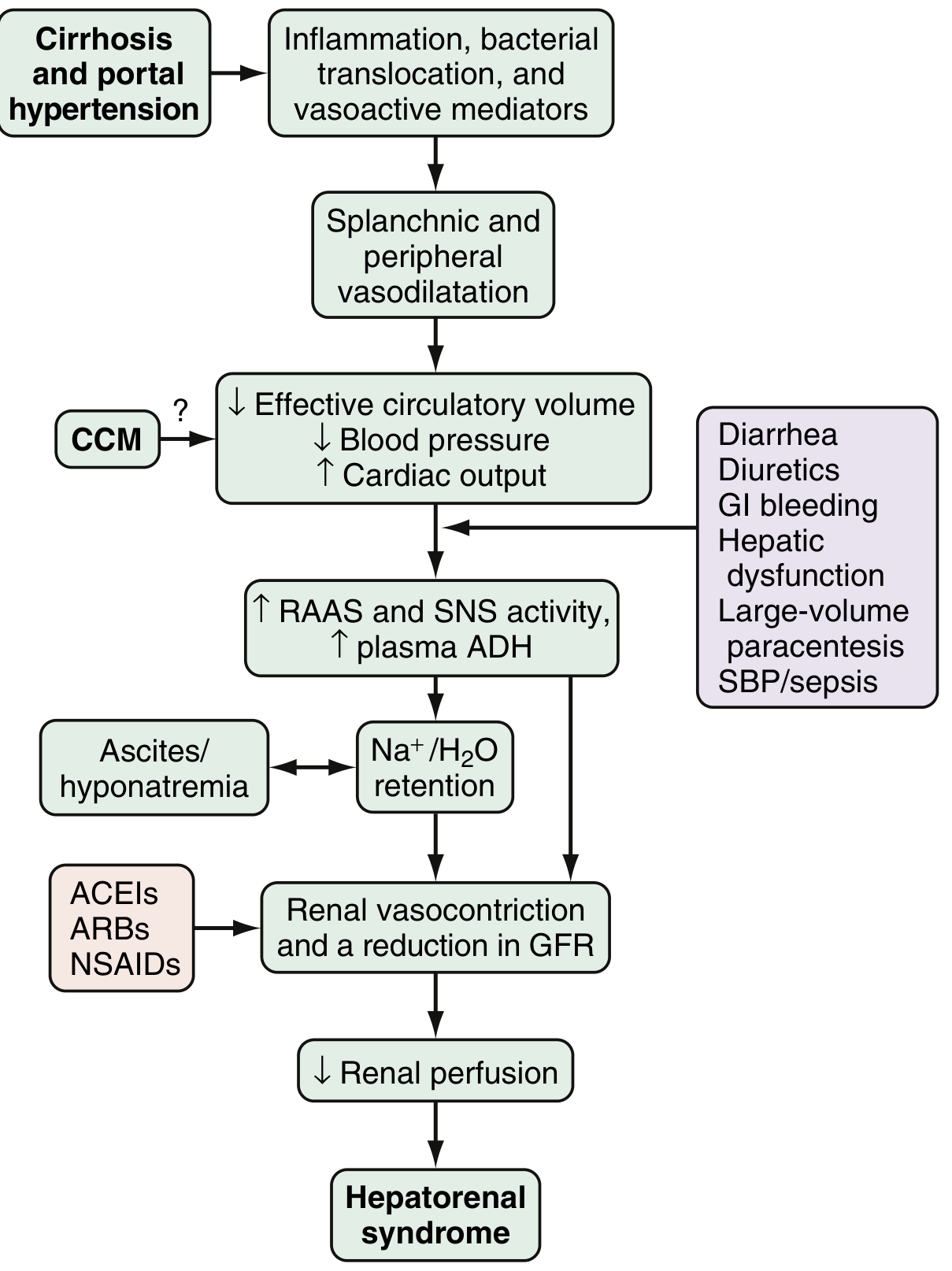
Fig. 94.2 from Sleisenger & Fordtran's - Proposed pathophysiology and triggers of HRS
Component 1: Splanchnic and Systemic Arterial Vasodilation
- Portal hypertension drives release of vasodilatory mediators: nitric oxide (NO), carbon monoxide, glucagon, prostacyclin, adrenomedullin, and endogenous opiates
- These cause intense splanchnic and peripheral vasodilation
- Effective circulating blood volume drops, blood pressure falls
- Early: increased heart rate and cardiac output compensate (hyperdynamic circulation)
- Late: compensatory mechanisms become exhausted
Component 2: Counter-regulatory Vasoconstriction (Renal)
- The body perceives decreased effective circulatory volume and activates:
- Renin-angiotensin-aldosterone system (RAAS)
- Sympathetic nervous system (SNS)
- Non-osmotic release of ADH/vasopressin
- These counteract systemic vasodilation but cause paradoxical renal vasoconstriction
- Altered local renal mediators (endothelins, prostaglandins, kallikreins) also contribute
- Result: markedly increased renal vascular resistance, decreased renal perfusion, fall in GFR
- With prolonged intense vasoconstriction, tubular damage can evolve - HRS shifts from functional to organic disease
Component 3: Cardiac Dysfunction (Cirrhotic Cardiomyopathy - CCM)
- Chronic cirrhosis impairs cardiac contractile reserve
- CCM worsens the mismatch between systemic vasodilation and cardiac output
- Contributes to further reduction in effective arterial blood volume
Precipitating Triggers
| Trigger | Mechanism |
|---|---|
| Spontaneous bacterial peritonitis (SBP) | Systemic inflammation, cytokines worsen vasodilation |
| GI bleeding | Hypovolemia, reduces effective circulating volume |
| Large-volume paracentesis (without albumin) | Acute loss of plasma oncotic support |
| Diuretic overuse / diarrhea (lactulose) | Volume depletion |
| NSAIDs | Block prostaglandin-mediated renal vasodilation |
| ACEIs / ARBs | Reduce angiotensin II-mediated renal efferent arteriole tone |
(Sleisenger & Fordtran's; Comprehensive Clinical Nephrology, 7th Ed.)
3. Classification: HRS-AKI vs. HRS-CKD
The older Type 1 / Type 2 classification has been updated by the International Club of Ascites (ICA):
| Feature | HRS-AKI (formerly Type 1) | HRS-CKD (formerly Type 2) |
|---|---|---|
| Onset | Rapid, progressive | Gradual, stable |
| Creatinine rise | Stage 2-3 AKI (>2x baseline or >4 mg/dL) | Persistent elevation, slower course |
| GFR | Rapidly declining | Moderately reduced, relatively stable |
| Typical presentation | Acute decompensation, SBP, sepsis | Diuretic-refractory ascites |
| Oliguria | Universal | Less prominent |
| Associated features | Hepatic failure, encephalopathy, multi-organ dysfunction, adrenal insufficiency | Refractory ascites |
| Survival without treatment | 2-3 weeks | Months |
| Prognosis | Poor | Better than HRS-AKI, but still poor |
(Comprehensive Clinical Nephrology, 7th Ed.; Harrison's 22E)
4. Diagnostic Criteria (ICA 2015 Revised)
HRS is a diagnosis of exclusion. All six criteria must be met:
- Cirrhosis with ascites
- AKI by ICA-AKI criteria: Rise in serum creatinine ≥0.3 mg/dL within 48 h, OR ≥50% rise from baseline within 7 days
- No response after at least 2 days of: (a) diuretic withdrawal, AND (b) volume expansion with albumin 1 g/kg/day (max 100 g/day)
- Absence of shock
- No current or recent nephrotoxic drugs (NSAIDs, aminoglycosides, iodinated contrast)
- No parenchymal kidney disease: proteinuria <500 mg/day, no microhematuria (>50 RBC/hpf), normal renal ultrasound
AKI Staging in Cirrhosis
| Stage | Creatinine Criteria |
|---|---|
| Stage 1 | Rise ≥0.3 mg/dL OR 1.5-2x baseline |
| Stage 2 | Rise >2-3x baseline |
| Stage 3 | Rise >3x baseline OR creatinine ≥4 mg/dL with acute rise ≥0.3 mg/dL |
HRS-AKI corresponds to Stage 2 or 3.
Key caveat: Muscle mass and urea synthesis are reduced in cirrhotic patients - serum creatinine may be deceptively low. Even small creatinine rises carry significance and require prompt attention.
(Sleisenger & Fordtran's; Goldman-Cecil Medicine)
5. Clinical Features
- Often asymptomatic in early stages
- Decreased urine output (oliguria nearly universal in HRS-AKI)
- Worsening ascites (often refractory to diuretics)
- Hyponatremia (almost always present)
- Low arterial blood pressure
- Hepatic encephalopathy may be the first presenting sign
- Jaundice, coagulopathy (markers of underlying liver failure)
- Features of precipitating cause (e.g., SBP signs)
Urine findings (important for DDx):
- Urine sodium very low (<10 mEq/L) - sodium avid state
- Urine osmolality > plasma osmolality
- No proteinuria or hematuria (distinguishes from glomerular disease)
- Bland urinary sediment
6. Differential Diagnosis of AKI in Cirrhosis
| Category | Examples |
|---|---|
| Pre-renal (hypovolemia) | Over-diuresis, GI bleeding, diarrhea |
| HRS | Functional, diagnosis of exclusion |
| Acute tubular necrosis (ATN) | Sepsis, aminoglycosides, contrast |
| Glomerulonephritis | IgA nephropathy (alcohol-related cirrhosis), hepatitis B/C-related GN |
| Obstructive | Rare in cirrhosis |
| Drug-induced | NSAIDs (inhibit prostaglandin renal perfusion), ACEIs, ARBs |
Urinary biomarkers in development (not yet standard): IL-18, NGAL (neutrophil gelatinase-associated lipocalin), EGF, FABP2 - may help distinguish HRS from ATN, though role in clinical practice not yet well-defined.
(Goldman-Cecil Medicine; Sleisenger & Fordtran's)
7. Epidemiology and Risk Factors
Independent predictors of developing HRS in cirrhotics:
- Dilutional hyponatremia
- Impaired systemic hemodynamics: high plasma renin activity, elevated noradrenaline, low cardiac output
- Abnormal renal duplex Doppler ultrasound: resistive index >0.7
- Low GFR at baseline
Incidence: 18% at 1 year, 39% at 5 years in cirrhotics. Neither Child-Pugh score nor MELD score predicts onset.
(Comprehensive Clinical Nephrology, 7th Ed.)
8. Prevention
Prevention is the most effective strategy given the high mortality once HRS is established:
- Avoid intravascular volume depletion: judicious diuretic use, correct diarrhea from lactulose promptly, always give albumin with large-volume paracentesis
- Large-volume paracentesis: always give IV albumin 6-8 g per liter of ascites removed (prevents post-paracentesis circulatory dysfunction)
- SBP: administer IV albumin (1.5 g/kg on day 1, 1 g/kg on day 3) - reduces HRS development and improves survival
- Norfloxacin prophylaxis: in patients with low ascitic protein (<1.5 g/dL) and either Child-Pugh ≥9 with bilirubin ≥3 mg/dL, or renal impairment (creatinine ≥1.2, BUN ≥25, or Na ≤130) - reduces HRS risk
- Pentoxifylline in severe alcoholic hepatitis (reduces TNF-alpha mediated renal vasoconstriction)
- Avoid nephrotoxins: NSAIDs, ACEIs, ARBs, aminoglycosides
- Treat infections promptly: SBP, bacteremia, sepsis
(Sleisenger & Fordtran's Box 94.3)
9. Treatment
Step 1: General Measures (All Patients)
- Stop all nephrotoxic agents: diuretics, NSAIDs, ACEIs, ARBs, aminoglycosides
- Treat infections: antibiotics for SBP, sepsis
- IV albumin bolus: 1 g/kg/day (max 100 g/day) for minimum 2 days
- If no response after 48 h of albumin + diuretic withdrawal, HRS-AKI diagnosis is confirmed
- Continue albumin at 20-60 g/day to maintain CVP 10-15 cm H₂O
Step 2: Vasoconstrictor Therapy
The key principle: reverse splanchnic vasodilation to offload the renal vasoconstriction. Always combine with albumin.
First-line: Terlipressin + Albumin (preferred where available)
- Selective vasopressin V1 receptor agonist; approved in Europe; FDA-approved in the USA since 2022
- Dosing: Start 1 mg IV every 4 hr; increase to 2 mg IV every 4 hr if creatinine does not fall ≥25% by day 3
- Also can be given as continuous IV infusion (better tolerated, requires lower doses)
- Efficacy: reverses HRS in 27-43% vs. 8-13% with albumin alone (OT-0401 and REVERSE trials)
- Best response in patients with lower baseline creatinine and lower bilirubin - supports early initiation
- Side effects: cardiovascular complications (ischemia), fluid overload - requires close monitoring
- Duration: maximum 2 weeks, until HRS reverses or LT is performed
Second-line: Norepinephrine + Albumin (ICU, widely available)
- Dosing: 0.1-0.7 μg/kg/min IV infusion; increase by 0.05 μg/kg/min every 4 hr; titrate to MAP increase of ≥10 mmHg
- Efficacy comparable to terlipressin in small RCTs; requires ICU monitoring
- Advantage: widely available and less expensive
Third-line: Midodrine + Octreotide + Albumin (outpatient feasible)
- Used commonly in the USA (historically, before terlipressin approval); now considered third-line
- Midodrine (α₁-adrenergic agonist, oral): start 2.5-5 mg PO TID; increase to max 15 mg TID; titrate to MAP increase ≥15 mmHg
- Octreotide (somatostatin analog - inhibits endogenous vasodilators): 100 μg SC TID to max 200 μg SC TID; or 25 μg IV bolus then 25 μg/hr infusion
- In head-to-head RCT: terlipressin was significantly superior (70.4% recovery vs. 28.6%)
Step 3: Renal Replacement Therapy (RRT)
- If medical treatment fails, RRT (hemodialysis) can be initiated as a bridge to liver transplantation
- Not a definitive therapy; does not address the underlying hepatic dysfunction
- Decision to initiate must weigh transplant candidacy and prognosis
Step 4: TIPS (Transjugular Intrahepatic Portosystemic Shunt)
- Reduces portal hypertension and, secondarily, splanchnic vasodilation
- May improve renal function in carefully selected patients
- Limited evidence; primarily a bridge to LT
- Contraindicated in severe hepatic encephalopathy, advanced hepatic failure (bilirubin >5 mg/dL, Child-Pugh >13)
Step 5: Liver Transplantation (Definitive Therapy)
- The only cure for HRS
- Recovery of renal function is typical after LT
- Patients with combined HRS-AKI or HRS-CKD have poor prognosis unless transplant is achieved
- MELD score incorporates creatinine, bilirubin, and INR - patients with HRS naturally receive high MELD scores, prioritizing them for LT
- Simultaneous liver-kidney transplantation (SLKT) considered when renal recovery after LT is unlikely (prolonged AKI >4-6 weeks)
(Sleisenger & Fordtran's Box 94.3; Harrison's 22E; Comprehensive Clinical Nephrology, 7th Ed.)
10. Prognosis
| Type | Median Survival (No Treatment) |
|---|---|
| HRS-AKI (Type 1) | 2-3 weeks |
| HRS-CKD (Type 2) | Months (better but still poor) |
- With vasoconstrictor treatment: reversal in 27-43% (terlipressin)
- Even after reversal, recurrence is high unless LT is performed
- Liver transplantation markedly improves survival; renal function typically recovers
- The kidney, when transplanted into a non-cirrhotic recipient, functions normally - confirming the purely functional nature of HRS
11. Recent Evidence (2024-2026)
A 2026 meta-analysis (PMID 41853888) compared continuous vs. bolus terlipressin for HRS and acute variceal bleeding in cirrhosis, adding to the evidence that continuous infusion is better tolerated than bolus dosing for HRS. No recent meta-analysis has changed the core management framework, and the primary algorithm (terlipressin + albumin as first-line, LT as definitive therapy) remains supported.
12. Quick Clinical Summary
Suspect HRS when:
Cirrhotic patient + ascites + rising creatinine
Rule out:
Volume depletion → Give albumin 1 g/kg/day x 2 days
Nephrotoxins → Stop diuretics, NSAIDs, ACEIs, ARBs
Infection → Check ascites (SBP), blood cultures
ATN/glomerular disease → Urine sediment, protein, imaging
If no response to albumin challenge → HRS-AKI confirmed
Treat:
1st: Terlipressin + albumin
2nd: Norepinephrine + albumin (ICU)
3rd: Midodrine + Octreotide + albumin
Bridge: RRT, TIPS
Definitive: Liver Transplantation
Prevent:
Albumin with LVP, treat SBP with albumin,
norfloxacin prophylaxis in high-risk patients,
avoid nephrotoxins
Sources: Harrison's Principles of Internal Medicine 22E (2025) - Sleisenger and Fordtran's Gastrointestinal and Liver Disease - Comprehensive Clinical Nephrology, 7th Edition - Goldman-Cecil Medicine
This is a shared conversation. Sign in to Orris to start your own chat.