Ischemic Heart Disease ( General Medicine ).
"ischemic heart disease" AND "management"
ischemic heart disease coronary atherosclerosis myocardial infarction diagram
Ischemic Heart Disease (IHD)
Definition
Epidemiology
- Leading cause of death in higher-income nations; accounts for ~7.5 million deaths worldwide per year
- ~800,000 Americans experience an MI annually; roughly half die
- IHD mortality in the USA has declined ~50% since peaking in 1963 — attributed to smoking cessation, BP/DM control, statins, aspirin, CCUs, thrombolysis, PCI, and CABG
- The trend is threatened by aging populations and the global obesity epidemic
Pathogenesis
1. Atherosclerosis (Chronic Fixed Occlusion)
- Plaques occluding < 70% of coronary lumen → typically asymptomatic
- ≥ 70% stenosis ("critical stenosis") → symptomatic on exertion (stable angina)
- > 90% stenosis → may cause symptoms at rest
2. Acute Plaque Change + Thrombosis
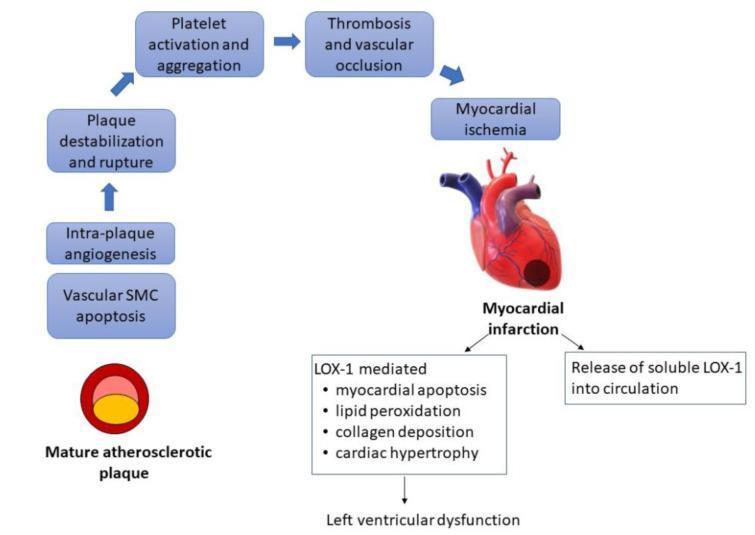
- Atheromatous plaque is eroded or disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces → subendothelial collagen and necrotic contents exposed
- Platelets adhere, aggregate, and activate → release thromboxane A₂, ADP, serotonin → further aggregation and vasospasm
- Coagulation activated by tissue factor exposure → thrombus grows
- Within minutes, thrombus may completely occlude the lumen
Angiography within 4 hours of MI shows thrombotic occlusion in ~90% of cases; by 12–24 hours only 60% even without intervention (some clear spontaneously).
3. Non-atherosclerotic Causes (minority)
- Increased demand: tachycardia, hypertension
- Reduced supply: hypotension/shock, anemia, CO poisoning, severe hypoxemia
Clinical Syndromes
| Syndrome | Key Features |
|---|---|
| Stable angina | Predictable exertional chest pain; fixed stenosis ≥70%; relieved by rest/nitrates |
| Unstable angina | Crescendo pattern; pain at rest or minimal exertion; non-occlusive thrombus |
| NSTEMI | Troponin positive; no complete occlusion; subendocardial infarction |
| STEMI | Complete occlusion; transmural infarction; urgent revascularization needed |
| Sudden cardiac death (SCD) | Fatal arrhythmia (usually VF); often first manifestation of IHD |
| Chronic IHD / Ischemic cardiomyopathy | Progressive CHF from accumulated ischemic injury |
Vasospastic (Prinzmetal) Angina
- Occurs at rest, often at night; due to coronary artery spasm (not fixed stenosis)
- ECG: transient ST elevation during pain
- Responds to nitrates and calcium channel blockers
Myocardial Response to Ischemia
| Time | Event |
|---|---|
| Seconds | Aerobic metabolism ceases; ATP drops; lactic acid accumulates |
| < 2 minutes | Loss of contractility |
| 20–40 minutes | Irreversible coagulative necrosis begins |
| > 20 min | Sarcolemmal integrity lost → intracellular macromolecules leak (basis for biomarkers) |
- If flow restored before irreversible injury: myocardium salvageable ("stunned myocardium" may be dysfunctional for days)
- Reperfusion injury: contraction bands, free radical damage (but reperfusion is still always net beneficial)
Morphological Changes in MI (Gross & Histological Timeline)
| Time After Infarction | Gross Changes | Microscopic Changes |
|---|---|---|
| 0–4 hours | None visible | None (or early wavy fibres) |
| 4–12 hours | Slight pallor | Coagulative necrosis beginning; oedema |
| 12–24 hours | Pallor, sometimes mottling | Coagulative necrosis; neutrophil infiltration begins |
| 1–3 days | Yellow-tan pallor; soft | Dense neutrophil infiltration |
| 3–7 days | Hyperaemic border; central yellow softening | Macrophages; necrotic debris removal |
| 1–2 weeks | Gelatinous, depressed | Granulation tissue ingrowth; prominent vasculature |
| 2–8 weeks | Fibrous (white) scar formation | Collagen deposition |
| > 2 months | Dense fibrous scar | Dense collagen scar |
Note: Reperfused MI appears haemorrhagic (bleeding from damaged vessels) with contraction bands microscopically — a hallmark of reperfusion injury.
Diagnosis
ECG
- STEMI: ST elevation in leads corresponding to infarcted territory; eventual Q waves (transmural)
- NSTEMI: ST depression, T-wave inversions; no Q waves
- Stable angina: normal at rest, may show ST depression on exercise stress test
Cardiac Biomarkers
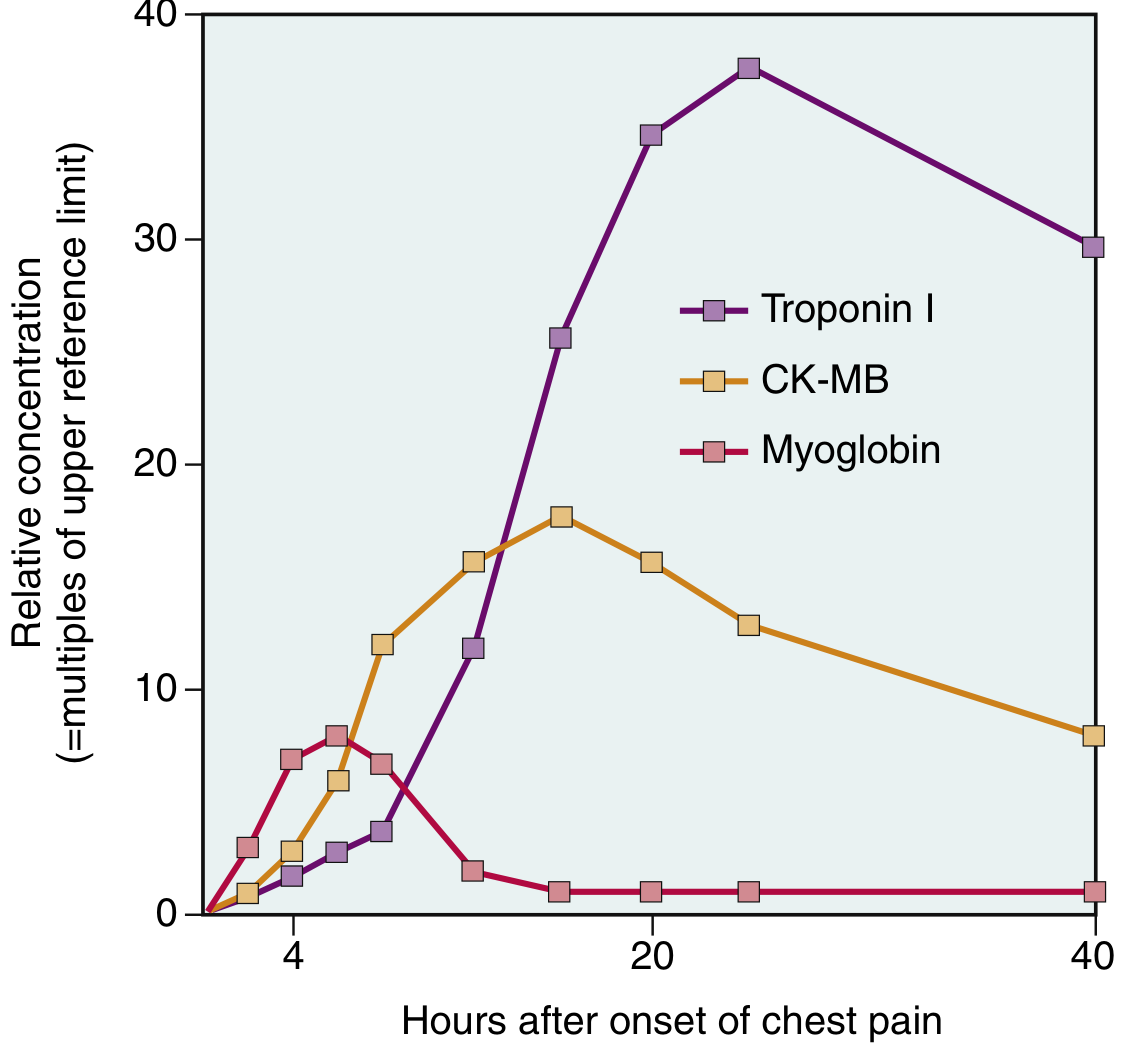
| Biomarker | Rises | Peaks | Returns to Normal | Notes |
|---|---|---|---|---|
| Myoglobin | 1–2 hr | 4–6 hr | ~24 hr | First to rise; poor specificity |
| CK-MB | 2–4 hr | 24–48 hr | ~72 hr | Good for re-infarction |
| Troponin I/T | 2–4 hr | ~24 hr | 7–14 days | Gold standard; highest sensitivity & specificity |
Imaging & Other Tests
- ECG stress test: first-line for stable CAD evaluation
- Coronary CTA: anatomical; excellent negative predictive value (SCOTT-Heart trial: associated with reduced MI at 5 years)
- Echocardiography: wall motion abnormalities; LV function (EF)
- Radionuclide perfusion (SPECT/PET): functional ischemia
- Coronary angiography: gold standard; allows simultaneous intervention
Management
Stable Angina / Chronic CAD
- Antiplatelet: Aspirin 75–100 mg/day (cornerstone)
- Beta-blockers: reduce myocardial O₂ demand; first-line for angina
- Nitrates: sublingual GTN for acute relief; long-acting for prophylaxis
- Calcium channel blockers: amlodipine/diltiazem — especially for vasospastic angina
- Statins: LDL-lowering; plaque stabilization (target LDL < 1.8 mmol/L for high risk)
- ACE inhibitors/ARBs: reduce cardiovascular events, especially if LV dysfunction or DM
- PCI (Percutaneous Coronary Intervention): balloon angioplasty + drug-eluting stent; preferred for single/double vessel disease
- CABG (Coronary Artery Bypass Grafting): preferred for left main disease, triple-vessel disease, LV dysfunction (EF < 35%), or diabetes with multi-vessel disease
ACS (NSTEMI / Unstable Angina)
- Dual antiplatelet therapy (DAPT): Aspirin + P2Y₁₂ inhibitor (clopidogrel, ticagrelor, or prasugrel)
- Anticoagulation: LMWH (enoxaparin) or fondaparinux or UFH
- Beta-blocker, statin, ACE inhibitor
- Early invasive strategy (angiography within 24–72 h) for high-risk NSTEMI (GRACE score, troponin positive, dynamic ECG changes)
STEMI — Emergency Management
Time is myocardium — aim: Door-to-balloon time < 90 minutes
- Primary PCI — treatment of choice; superior to thrombolysis if available within 90–120 min
- Thrombolysis (alteplase, tenecteplase) if PCI unavailable within 120 min; contraindicated in recent surgery, stroke, active bleeding
- Aspirin + P2Y₁₂ inhibitor + anticoagulation
- IV beta-blocker (if haemodynamically stable)
- Nitrates (not in inferior MI with RV involvement — may drop BP)
- Morphine for pain
- High-intensity statin
- DAPT for 12 months, then aspirin lifelong
- ACE inhibitor (especially if EF < 40%)
- Beta-blocker
- Aldosterone antagonist (eplerenone) if EF < 35% + symptoms of HF
- Cardiac rehabilitation
- ICD implantation if EF < 35% after ≥ 40 days
Complications of MI
| Complication | Timing | Features |
|---|---|---|
| Arrhythmias | Immediate (hours) | Most common cause of pre-hospital death; VF, VT, heart block |
| LV failure / pulmonary oedema | Hours–days | Impaired contractility; treat with diuretics, ACE-I |
| Cardiogenic shock | Hours–days | > 40% LV involved; high mortality; IAB pump / Impella |
| Free wall rupture | Days 3–7 | Haemopericardium → tamponade; usually fatal |
| Interventricular septal defect (VSD) | Days 3–7 | New loud harsh systolic murmur; confirmed by echo |
| Papillary muscle rupture | Days 3–7 | Acute severe mitral regurgitation; pulmonary oedema; surgical emergency |
| Pericarditis (Dressler's syndrome) | 2–10 days (Dressler: weeks–months) | Pleuritic chest pain, fever, friction rub |
| Mural thrombus | Days–weeks | LV apex; risk of systemic embolism; anticoagulate |
| Ventricular aneurysm | Weeks–months | Persistent ST elevation; CHF; arrhythmia; thrombus |
| Chronic ischaemic cardiomyopathy | Months–years | Progressive HF from scar and remodelling |
Risk Factors
- Hypertension (most important)
- Dyslipidaemia (elevated LDL, low HDL)
- Diabetes mellitus
- Smoking
- Obesity / metabolic syndrome
- Physical inactivity
- Diet (saturated fat, trans fats)
- Psychosocial stress
Key Points Summary
- IHD results from coronary perfusion-demand mismatch; atherosclerosis is responsible in > 90%
- Plaque rupture + thrombosis is the trigger for most acute events (MI, SCD)
- Irreversible myocyte necrosis occurs after 20–40 minutes of ischemia
- Troponin I/T are the gold-standard biomarkers; rise 2–4h, peak ~24h
- ACS = unstable angina + NSTEMI + STEMI; each requires risk stratification and early treatment
- STEMI demands primary PCI within 90 minutes (door-to-balloon)
- Post-MI therapy: DAPT, beta-blocker, ACE-I, statin, cardiac rehab; ICD if EF < 35%
- Major complications: arrhythmia (immediate), mechanical rupture (days 3–7), aneurysm/CHF (late)
Risk Factors for Atherosclerosis
"atherosclerosis" AND "risk factors"
atherosclerosis risk factors pathogenesis foam cells endothelial dysfunction
Hi
Atherosclerosis pathogenesis for writing in University exam final year MBBS
Pathogenesis of Atherosclerosis
Definition
The "Response to Injury" Hypothesis (Contemporary/Accepted Theory)
Step-by-Step Pathogenesis
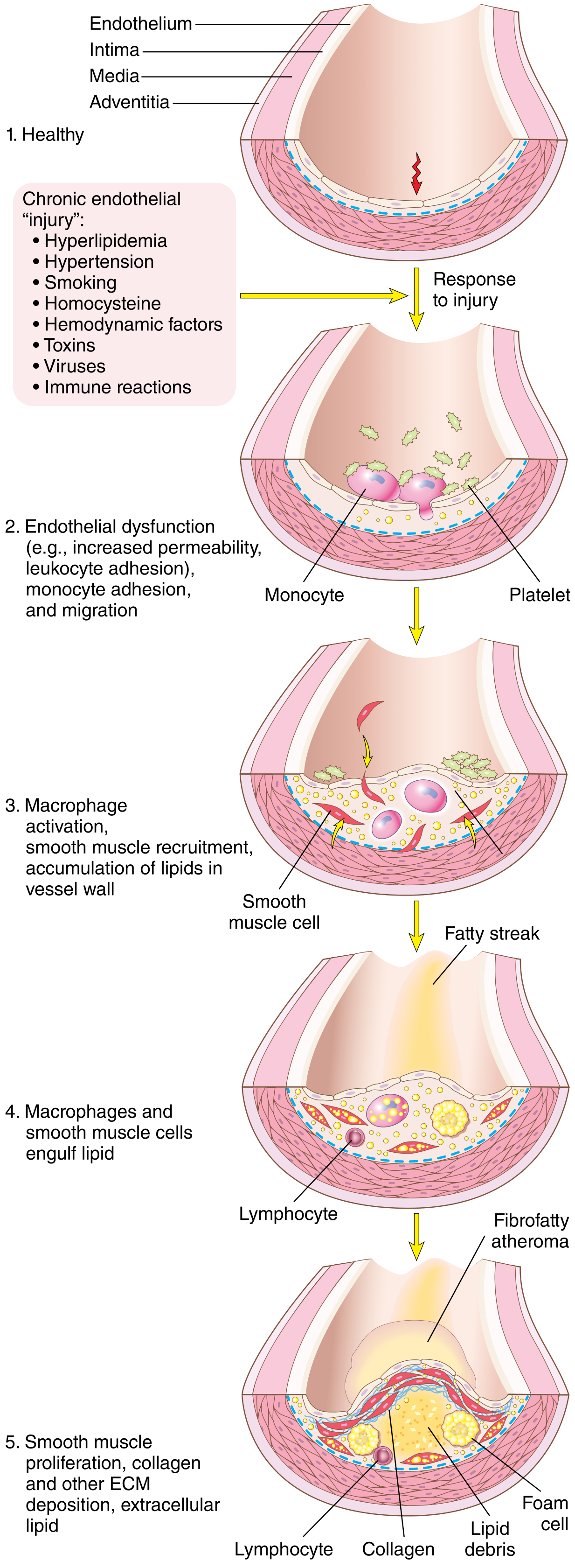
STEP 1 — Endothelial Injury and Dysfunction
- Increased vascular permeability
- Enhanced leukocyte adhesion (upregulation of VCAM-1, ICAM-1, E-selectin)
- Altered (pro-inflammatory) gene expression
- Reduced nitric oxide (NO) production → loss of vasodilation and anti-inflammatory signalling
- Hemodynamic disturbances (turbulent, non-laminar blood flow) — explains why plaques localise at branch points, ostia of vessels, and the posterior abdominal aorta. Laminar flow induces the transcription factor KLF-2, which turns on atheroprotective genes; turbulent flow suppresses this.
- Hypercholesterolaemia — most important biochemical cause
- Hypertension — mechanical shear stress
- Cigarette smoke toxins
- Inflammatory cytokines (e.g., TNF-α)
- Homocysteinaemia, infections, immune reactions
STEP 2 — Lipoprotein Accumulation and Oxidation
- LDL cholesterol accumulates in the intima (subendothelial space) due to increased EC permeability
- LDL is oxidised by oxygen free radicals generated locally by macrophages and ECs → oxidised LDL (oxLDL)
- Chronic hyperlipidaemia accelerates NO decay (via free radical production), further impairing vasodilation
- oxLDL is cytotoxic to ECs and SMCs, and is a powerful pro-inflammatory stimulus
- Cholesterol crystals also form, contributing to local inflammation
Evidence for lipid role: Plaques consist predominantly of cholesterol; familial hypercholesterolaemia (defective LDL receptors) causes MI by age 20; lowering LDL with statins slows plaque progression and reduces cardiovascular events.
STEP 3 — Monocyte Recruitment and Foam Cell Formation
- Dysfunctional ECs express adhesion molecules (VCAM-1) → circulating monocytes adhere to the endothelium
- Monocytes transmigrate into the intima, driven by chemokines (especially MCP-1)
- In the intima, monocytes differentiate into macrophages
- Macrophages engulf oxLDL via scavenger receptors (bypassing normal LDL receptor regulation — unregulated uptake)
- Lipid-laden macrophages become foam cells
- Accumulation of foam cells produces the earliest grossly visible lesion: the fatty streak (yellow, flat, intimal streak)
STEP 4 — Platelet Adhesion and Activation
- EC injury → platelets adhere to exposed subendothelial collagen
- Activated platelets release:
- PDGF (Platelet-Derived Growth Factor) → stimulates SMC migration and proliferation
- TGF-β → promotes collagen synthesis
- Thromboxane A₂ → vasoconstriction and further platelet aggregation
STEP 5 — Smooth Muscle Cell (SMC) Proliferation and ECM Production
- Growth factors from platelets, macrophages, and ECs (especially PDGF, FGF, TGF-β) recruit SMCs from the media into the intima
- SMCs undergo phenotypic change from contractile to synthetic type
- Intimal SMCs proliferate and synthesise extracellular matrix (ECM) — collagen, proteoglycans, elastin
- This forms the fibrous cap overlying the lipid core
- T lymphocytes are also recruited → release cytokines (IFN-γ) that further activate macrophages
STEP 6 — Plaque Development (Fibrofatty Atheroma)
| Component | Origin |
|---|---|
| Fibrous cap | Collagen + proteoglycans synthesised by SMCs |
| Lipid core | Cholesterol, cholesterol esters, necrotic debris |
| Foam cells | Lipid-laden macrophages |
| T lymphocytes | Inflammatory cells |
| Calcification | Late stage — calcium deposits in necrotic core |
| Neovascularisation | New capillaries from adventitia |
STEP 7 — Plaque Complication and Rupture
- Thin fibrous cap
- Large lipid/necrotic core
- Dense inflammatory infiltrate (macrophages + T cells)
- Few SMCs (reduced collagen synthesis)
- Macrophages secrete matrix metalloproteinases (MMPs) → collagen degradation → cap thinning
- IFN-γ (from T cells) inhibits SMC collagen synthesis
- Net effect: collagen breakdown > synthesis → mechanically weak cap
- Plaque rupture → thrombogenic plaque contents exposed → platelet aggregation + thrombus → complete occlusion → STEMI / MI
- Plaque erosion/ulceration → subendothelial basement membrane exposed → partial thrombosis → NSTEMI / unstable angina
- Intraplaque haemorrhage → sudden expansion of plaque volume → rapid luminal narrowing
- Embolisation → distal microinfarcts
Important: Most plaques that rupture cause acute MI had only mild-to-moderate stenosis (not "critical" stenosis) before rupture — the degree of stenosis does not predict rupture risk; plaque composition does.
Morphological Progression (Summary)
Normal intima
↓
Fatty streak (foam cells, earliest lesion, seen even in young children)
↓
Intermediate lesion (more lipid + SMCs)
↓
Fibrofatty plaque (fibrous cap + lipid core) → stable angina when >70% stenosis
↓
Complicated plaque (rupture / calcification / haemorrhage / thrombosis)
↓
Acute coronary syndrome / Stroke / Peripheral vascular disease
Role of Inflammation — Key Exam Point
- C-reactive protein (CRP) — elevated even in patients without hyperlipidaemia; adds prognostic information above Framingham risk score
- Statin drugs reduce cardiovascular risk partly through anti-inflammatory effects (independent of LDL-lowering), including upregulation of KLF-2
- Inflammatory cells (macrophages, T cells) are present throughout plaque evolution
- Clonal haematopoiesis of indeterminate potential (CHIP) — mutated monocyte clones with pro-inflammatory phenotype markedly increase atherosclerosis risk with aging
Key Points for Exam
| Point | Detail |
|---|---|
| Theory | "Response to Injury" — chronic inflammatory healing response |
| Cornerstone | Endothelial injury/dysfunction |
| Most important causes | Hemodynamic turbulence + hypercholesterolaemia |
| Key lipid | oxLDL → foam cells via scavenger receptors |
| Foam cells | Lipid-laden macrophages — form fatty streak |
| Fibrous cap made by | Smooth muscle cells (collagen synthesis) |
| Cap degraded by | MMPs from macrophages |
| Vulnerable plaque | Thin cap + large necrotic core + inflammation |
| Plaque rupture leads to | Thrombosis → MI / stroke |
| Atheroprone sites | Branch points, ostia, posterior abdominal aorta |
| Atheroprotection | Laminar flow → KLF-2 → protective gene expression |
For general medicine paper
ATHEROSCLEROSIS — Pathogenesis
For General Medicine Final Year MBBS Exam
INTRODUCTION
THEORIES OF PATHOGENESIS
| Theory | Proponent | Concept |
|---|---|---|
| Lipid/Insudation theory | Virchow | Lipid accumulates passively in the intima |
| Thrombogenic theory | Rokitansky | Mural thrombi organise to form plaques |
| Response to Injury hypothesis | Ross (1976, revised 1999) | Currently accepted — chronic inflammatory response to endothelial injury |
THE "RESPONSE TO INJURY" HYPOTHESIS (Current Accepted Theory)
"Atherosclerosis is a chronic inflammatory and healing response of the arterial wall to endothelial injury." — Robbins, Cotran & Kumar
STEP-BY-STEP PATHOGENESIS
Stage 1 — Endothelial Injury and Dysfunction
- ↑ Vascular permeability
- ↑ Leukocyte adhesion (upregulation of VCAM-1, ICAM-1, selectins)
- ↓ Nitric oxide (NO) production → loss of vasodilatory and anti-inflammatory protection
- Pro-thrombotic tendency
- Hemodynamic turbulence — most important physical cause
- Hypercholesterolaemia — most important biochemical cause
- Hypertension, cigarette smoke toxins, diabetes, homocysteinaemia, inflammatory cytokines (TNF-α)
Atheroprone sites: Branch points, ostia of vessels, posterior abdominal aorta — all areas of turbulent, non-laminar flow. Laminar flow induces the transcription factor KLF-2 which turns on atheroprotective genes; turbulence suppresses KLF-2, making these sites prone to atherogenesis.
Stage 2 — Lipoprotein Accumulation and Oxidation
- ↑ EC permeability → LDL accumulates in the intima
- LDL is oxidised by oxygen free radicals from macrophages and ECs → oxidised LDL (oxLDL)
- oxLDL is central to atherogenesis because it:
- Is cytotoxic to ECs and SMCs
- Stimulates release of chemokines (MCP-1) attracting more monocytes
- Upregulates adhesion molecules on ECs
- Accelerates NO decay → further endothelial dysfunction
- Promotes foam cell formation via macrophage scavenger receptors
Stage 3 — Monocyte Adhesion, Migration, and Foam Cell Formation
- Dysfunctional ECs express VCAM-1 and selectins → circulating monocytes adhere
- Monocytes transmigrate into the intima driven by chemokine MCP-1 (Monocyte Chemotactic Protein-1)
- In the intima → monocytes differentiate into macrophages
- Macrophages engulf oxLDL via scavenger receptors (CD36, SR-A) — this is unregulated (unlike normal LDL receptor-mediated uptake which is subject to negative feedback)
- Lipid-laden macrophages = Foam cells
- Accumulation of foam cells → Fatty streak — the earliest visible lesion of atherosclerosis (yellow, flat intimal streaks; seen even in aortas of children)
Stage 4 — Platelet Adhesion and Activation
- EC injury exposes subendothelial collagen → platelet adhesion and activation
- Activated platelets release:
- PDGF (Platelet-Derived Growth Factor) → stimulates SMC proliferation and migration
- TGF-β → promotes collagen and ECM synthesis
- Thromboxane A₂ → vasoconstriction, further platelet aggregation
Stage 5 — Smooth Muscle Cell (SMC) Migration and Proliferation
- Growth factors (PDGF, FGF, TGF-β) from platelets, macrophages, and ECs recruit SMCs from the media into the intima
- SMCs undergo phenotypic change: contractile → synthetic phenotype
- Intimal SMCs proliferate and synthesise collagen, elastin, proteoglycans → forming the fibrous cap
- T lymphocytes are also recruited → release IFN-γ activating macrophages further
Stage 6 — Formation of the Atherosclerotic Plaque
| Component | Detail |
|---|---|
| Fibrous cap | Dense collagen + SMCs (caps the lesion toward the lumen) |
| Lipid core | Cholesterol crystals, cholesterol esters, necrotic cell debris |
| Foam cells | Lipid-laden macrophages |
| Inflammatory infiltrate | Macrophages, T lymphocytes, mast cells |
| Neovascularisation | New capillaries from adventitia (prone to haemorrhage) |
| Calcification | Late-stage dystrophic calcification of necrotic core |
Glagov phenomenon: Early in plaque development, the vessel wall remodels outward (compensatory enlargement) to preserve lumen size. Eventually this compensation is exhausted → lumen compromise.Critical stenosis: ≥ 70–75% reduction in luminal cross-sectional area → sufficient to cause tissue ischaemia at rest or exertion.
Stage 7 — Plaque Complication and Rupture (Clinically Most Important)
- Thin fibrous cap
- Large lipid/necrotic core (> 40% of plaque volume)
- Dense inflammatory infiltrate (macrophages + T cells at the shoulder region)
- Few SMCs → reduced collagen synthesis → mechanically weak cap
- Macrophages secrete Matrix Metalloproteinases (MMPs) → degrade collagen
- T cells secrete IFN-γ → inhibit SMC collagen synthesis
- Net result: collagen degradation > synthesis → thin, rupture-prone cap
⚠️ Critical exam point: Most plaques that rupture and cause MI had only mild-to-moderate stenosis (< 70%) before rupture — the plaque composition matters more than its size. Standard angiography cannot predict which plaques will rupture.
| Event | Mechanism | Clinical Result |
|---|---|---|
| Plaque rupture | Cap tears; thrombogenic core exposed | Complete thrombosis → STEMI |
| Plaque erosion | Surface endothelium lost | Partial thrombosis → NSTEMI / Unstable angina |
| Intraplaque haemorrhage | Neovascular haemorrhage into plaque | Rapid plaque expansion → sudden stenosis |
| Atheroembolism | Plaque debris enters bloodstream | Microemboli → distal infarcts |
| Aneurysm | Media ischaemia → wall weakening | Aortic aneurysm, rupture |
SUMMARY FLOWCHART
Risk Factors (HTN, hyperlipidaemia, smoking, DM, etc.)
↓
Endothelial Injury / Dysfunction
↓
↑ Permeability, ↑ Adhesion molecules, ↓ NO
↓
LDL accumulation in intima → oxidised to oxLDL
↓
Monocyte adhesion & migration → Macrophages
↓
Macrophages engulf oxLDL (via scavenger receptors)
↓
Foam cells → FATTY STREAK (earliest lesion)
↓
Platelet adhesion → PDGF → SMC migration & proliferation
↓
Fibrous cap formation → FIBROFATTY PLAQUE
↓
MMP activity (macrophages) → cap thinning
↓
VULNERABLE PLAQUE → Rupture → Thrombosis
↓
ACUTE CORONARY SYNDROME / STROKE / PVD
CLINICAL CONSEQUENCES OF ATHEROSCLEROSIS
| Artery Involved | Clinical Manifestation |
|---|---|
| Coronary arteries | Angina, MI, sudden cardiac death |
| Carotid / cerebral arteries | TIA, ischaemic stroke |
| Aorta | Aortic aneurysm (usually abdominal) |
| Renal arteries | Renovascular hypertension, renal failure |
| Peripheral arteries | Intermittent claudication, gangrene |
| Mesenteric arteries | Mesenteric ischaemia, bowel infarction |
PREVENTION AND TREATMENT (Exam-relevant)
| Intervention | Target |
|---|---|
| Statins | ↓ LDL; plaque stabilisation via ↑ KLF-2, ↓ inflammation; ↓ MMP activity |
| Anti-hypertensives | Reduce hemodynamic injury |
| Antiplatelet agents (Aspirin) | Inhibit platelet aggregation at rupture sites |
| Smoking cessation | Removes major EC toxin |
| Glycaemic control | Reduces oxLDL formation and endothelial AGE deposition |
| PCSK9 inhibitors | Powerfully reduce LDL |
HIGH-YIELD POINTS FOR THE EXAM
- Theory = "Response to Injury" — chronic inflammatory response
- Cornerstone = Endothelial injury/dysfunction
- Earliest lesion = Fatty streak (foam cells only; reversible)
- Foam cells = oxLDL-laden macrophages via scavenger receptors
- Fibrous cap = Made by smooth muscle cells (collagen synthesis)
- Cap degradation = MMPs from macrophages + IFN-γ inhibiting SMC collagen synthesis
- Atheroprone sites = Areas of turbulent flow (branch points, ostia)
- Critical stenosis = ≥ 70–75% luminal narrowing
- Plaque rupture → Thrombosis → ACS (most plaques that rupture were < 70% stenosis)
- CRP = Marker of inflammation; adds prognostic information beyond Framingham risk score
Harrison reference
Atherosclerosis — Pathogenesis
Harrison's Principles of Internal Medicine, 22nd Edition (2025)
DEFINITION
"Epicardial coronary arteries are the major site of atherosclerotic disease." — Harrison's Principles of Internal Medicine, 22e, Chapter 285
HARRISON'S PERSPECTIVE ON PATHOGENESIS
STEP-BY-STEP PATHOGENESIS
1. Endothelial Dysfunction — The Initial Event
- Local control of vascular tone
- Maintenance of an anti-thrombotic surface
- Control of inflammatory cell adhesion and diapedesis
- Inappropriate vasoconstriction
- Luminal thrombus formation
- Abnormal monocyte and platelet interaction with the activated endothelium
"The loss of these defenses leads to inappropriate constriction, luminal thrombus formation, and abnormal interactions between blood cells, especially monocytes and platelets, and the activated vascular endothelium." — Harrison's 22e, Ch. 285 — Coronary Atherosclerosis
2. Lipoprotein Accumulation → Foam Cell Formation
- LDL enters the dysfunctional intima and becomes oxidised → oxLDL
- oxLDL triggers release of chemokines and adhesion molecules promoting inflammation
- Monocytes are recruited → differentiate into macrophages
- Macrophages accumulate cholesterol → become foam cells (lipid-laden macrophages)
- Other activated macrophages release pro-inflammatory mediators: TNF, IL-1, oxygen/nitrogen free radicals, eicosanoids, and prothrombotic factors
3. Smooth Muscle Cell Proliferation → Plaque Formation
- Growth factors from macrophages, platelets, and ECs → SMC migration from media into intima
- SMCs proliferate and synthesise collagen, proteoglycans, elastin → forming the fibrous cap
- This results in subintimal collections of fat, smooth muscle cells, fibroblasts, and intercellular matrix = the atherosclerotic plaque
4. Progressive Stenosis
| Degree of Diameter Reduction | Effect |
|---|---|
| 50% stenosis | Limitation of ability to increase flow with demand → effort angina |
| ~80% stenosis | Blood flow reduced at rest; minor further decrease → dramatic flow reduction |
- When a stenosis is severe, distal resistance vessels maximally dilate to maintain flow
- A pressure gradient develops across the stenosis → post-stenotic pressure falls
- Myocardial blood flow becomes dependent on distal coronary pressure
- Ischaemia is precipitated by increased O₂ demand (exercise, stress, tachycardia)
"When the resistance vessels are maximally dilated, myocardial blood flow becomes dependent on the pressure in the coronary artery distal to the obstruction." — Harrison's 22e, Ch. 285
5. Plaque Rupture / Erosion → Acute Coronary Syndrome (Most Clinically Important)
"STEMI usually occurs when coronary blood flow decreases abruptly after a thrombotic occlusion of a coronary artery previously affected by atherosclerosis." — Harrison's 22e, Ch. 286 — STEMI
- STEMI occurs when the surface of an atherosclerotic plaque is disrupted (rupture or erosion), exposing contents to blood
- Plaques prone to disruption have:
- Rich lipid core
- Thin fibrous cap
- Expansive remodelling
- Neovascularisation (angiogenesis)
- Plaque haemorrhage
- Adventitial inflammation
- "Spotty" pattern of calcification
- Plaque disrupts → subendothelial content exposed
- Initial platelet monolayer forms at disruption site
- Agonists (collagen, ADP, epinephrine, serotonin) activate platelets → release thromboxane A₂ (vasoconstriction + further aggregation)
- Platelet GP IIb/IIIa receptor activates → binds fibrinogen → platelet cross-linking and aggregation
- Tissue factor exposed from damaged ECs → activates factors VII and X → prothrombin → thrombin → fibrinogen → fibrin
- Thrombus of platelet aggregates + fibrin strands occludes the artery → STEMI
Note: Slowly developing high-grade stenoses typically do not precipitate STEMI because collateral circulation develops over time. STEMI results from rapid thrombosis at a site of plaque disruption.
6. Collateral Circulation
- Chronic severe coronary narrowing → development of collateral vessels
- When well developed, collaterals can maintain myocardial viability at rest but not during increased demand
- If stenosis occurs slowly, smaller adjacent vessels enlarge to partially compensate
HARRISON'S RISK FACTORS FOR ATHEROSCLEROSIS
- Older age
- Diabetes mellitus
- Hypertension (most significant — target BP < 130/80 mmHg; SPRINT trial: SBP < 120 mmHg reduces MI/stroke by 43%)
- Tobacco smoking
- Elevated LDL cholesterol
- Low HDL cholesterol
- Lipoprotein (a) excess
- Obesity
- Hypertriglyceridaemia
- Hyperfibrinogenaemia
- Homocystinuria
- Physical inactivity
- Prior TIA/stroke (for cerebrovascular disease)
- Small, dense LDL particles + hypertriglyceridaemia + low HDL
- Associated with: ↑ fibrinogen, ↑ PAI-1 (plasminogen activator inhibitor-1), ↑ factor VII, platelet hyperactivity
- Creates both atherogenic and thrombotic risk
- Does not cause atherosclerosis
- Reflects ongoing inflammation that may accelerate the process
- JUPITER trial (Harrison's reference): Patients with elevated CRP but LDL < 130 mg/dL treated with rosuvastatin showed 51% reduction in primary stroke (HR 0.49)
ANTIATHEROSCLEROTIC THERAPY (Harrison's)
| Intervention | Mechanism |
|---|---|
| Statins | ↓ LDL; ↓ inflammation; ↑ KLF-2 (atheroprotective); plaque stabilisation |
| Antihypertensives (thiazides, ACE-I) | Reduce haemodynamic EC injury |
| Antiplatelet agents | Inhibit GP IIb/IIIa, thromboxane A₂; prevent thrombus at rupture site |
| Smoking cessation | Removes major source of EC toxins |
| Glycaemic control | Reduces vulnerable blood and EC injury |
SUMMARY — HARRISON'S KEY POINTS
| Point | Harrison's Statement |
|---|---|
| Commonest cause | Atherosclerosis of epicardial coronary arteries |
| Primary mechanism | Endothelial dysfunction → loss of vascular tone, antithrombotic surface, and inflammatory control |
| Lipid role | oxLDL → chemokine release → monocyte recruitment → foam cells |
| Plaque rupture trigger | Thin-capped plaque with rich lipid core ruptures → platelet activation → thrombosis |
| 50% stenosis | Limits flow increase with demand (effort angina) |
| 80% stenosis | Resting flow reduced |
| STEMI mechanism | Rapid coronary thrombosis at plaque disruption site |
| Key platelet receptor | GP IIb/IIIa (fibrinogen binding → aggregation) |
| Coagulation trigger | Tissue factor → VII + X → thrombin → fibrin |
| Hypertension target | < 130/80 mmHg (SPRINT trial evidence) |
| Statins beyond cholesterol | SPARCL trial (atorvastatin 80 mg), JUPITER trial (rosuvastatin + elevated CRP) |
Explain the ETIOPATHOGENESIS, Clinical features, INVESTIGATION and MANAGEMENT of Causes of left side chest pain- explain acute MI
ACUTE MYOCARDIAL INFARCTION (AMI)
Etiopathogenesis, Clinical Features, Investigations & Management
Final Year MBBS — General Medicine | Harrison's 22e
PART 1: LEFT-SIDED CHEST PAIN — CAUSES (Differential Diagnosis)
| System | Cause | Key Distinguishing Feature |
|---|---|---|
| Cardiac | Acute MI (STEMI/NSTEMI) | Central/left chest, crushing, radiation to arm/jaw, diaphoresis |
| Cardiac | Unstable Angina | Similar to MI but no troponin rise, relieved by nitrates |
| Cardiac | Stable Angina | Exertional, relieved by rest/GTN |
| Cardiac | Acute Pericarditis | Sharp, pleuritic, relieved leaning forward, friction rub |
| Pulmonary | Pulmonary Embolism | Pleuritic, sudden onset, dyspnoea, hypoxia, risk factors |
| Pulmonary | Pneumothorax | Sudden, pleuritic, absent breath sounds |
| Pulmonary | Pneumonia/Pleuritis | Fever, productive cough, pleuritic |
| Vascular | Aortic Dissection | Tearing/ripping, radiates to back, BP differential between arms |
| GI | GERD / Oesophageal spasm | Burning, postprandial, relieved by antacids |
| GI | Peptic ulcer / Gastritis | Epigastric, related to meals |
| Musculoskeletal | Costochondritis | Localised, reproducible on palpation (Tietze's) |
| Neurological | Herpes Zoster | Dermatomal distribution, vesicular rash |
"The pain of STEMI can simulate pain from acute pericarditis, pulmonary embolism, acute aortic dissection, costochondritis, and gastrointestinal disorders." — Harrison's 22e
ACUTE MYOCARDIAL INFARCTION (AMI)
DEFINITION
MI = acute myocardial injury + clinical evidence of ischaemia + rise and/or fall of cardiac troponin (cTn) with at least one value above 99th percentile URL + at least ONE of:
- Symptoms of ischaemia
- New ischaemic ECG changes
- New pathological Q waves
- Imaging evidence of new wall motion abnormality / loss of viable myocardium
- Coronary thrombus on angiography/autopsy
PART 2: ETIOPATHOGENESIS
Types of MI (Harrison's Classification)
| Type | Mechanism |
|---|---|
| Type 1 | Acute atherothrombosis (plaque rupture/erosion) — most common |
| Type 2 | Supply-demand mismatch unrelated to thrombosis (e.g., vasospasm, anaemia, tachycardia) |
| Type 3 | Cardiac death with suspected MI before biomarkers available |
| Type 4a | PCI-related MI |
| Type 5 | CABG-related MI |
Pathogenesis of Type 1 MI (Plaque Rupture → Thrombosis)
- Atherosclerotic plaque with rich lipid core + thin fibrous cap ruptures or erodes
- Rupture-prone features: expansive remodelling, neovascularisation, plaque haemorrhage, adventitial inflammation, "spotty" calcification
- Injury facilitated by: cigarette smoking, hypertension, lipid accumulation, inflammation
- Plaque contents exposed to blood → initial platelet monolayer at rupture site
- Agonists (collagen, ADP, epinephrine, serotonin) → platelet activation
- Release of thromboxane A₂ → potent vasoconstriction + further platelet activation
- GP IIb/IIIa receptor conformation change → binds fibrinogen → platelet cross-linking and aggregation
- Tissue factor exposed from damaged ECs → activates Factors VII and X
- Prothrombin → Thrombin → Fibrinogen → Fibrin
- Thrombin autoamplifies the cascade
- Culprit artery occluded by platelet aggregates + fibrin strands = coronary thrombus
- Complete occlusion → cessation of blood flow → ischaemia → infarction
- Irreversible necrosis begins after 20–40 minutes of sustained ischaemia
- Territory supplied by affected vessel
- Whether occlusion is complete
- Duration of coronary occlusion
- Collateral blood supply
- Myocardial oxygen demand at time of occlusion
- Spontaneous thrombus lysis
- Adequacy of reperfusion when restored
Important: Slowly developing high-grade stenoses usually do NOT precipitate STEMI (collaterals develop); STEMI results from rapid thrombosis at a site of acute plaque disruption.
Precipitating Factors (Harrison's)
- Vigorous physical exercise
- Emotional stress
- Medical/surgical illness
- Circadian pattern — clusters in morning hours within hours of awakening (adrenergic surge)
Rare Causes of AMI (Non-Atherosclerotic)
- Coronary emboli
- Congenital coronary abnormalities
- Coronary artery spasm (vasospasm)
- Spontaneous coronary artery dissection (SCAD)
- Cocaine abuse
- Hypercoagulable states / collagen vascular disease
PART 3: CLINICAL FEATURES
Symptoms
- Deep and visceral in quality
- Described as: heavy, squeezing, crushing (occasionally stabbing or burning)
- Location: Central chest and/or epigastrium
- Radiation: Left arm, left shoulder, neck, jaw (less commonly: back, abdomen, occipital area — never below umbilicus)
- Duration: > 30 minutes (does NOT subside with rest or GTN — unlike angina)
- Onset: Often at rest; if during exertion, does NOT resolve with cessation of activity
- Diaphoresis (profuse sweating — highly suggestive)
- Nausea and vomiting
- Anxiety and sense of impending doom
- Dyspnoea (from LV dysfunction / pulmonary oedema)
- Palpitations (arrhythmias)
- Syncope / sudden cardiac death (from VF)
"The combination of substernal chest pain persisting for > 30 min and diaphoresis strongly suggests STEMI." — Harrison's 22e
- Diabetics (autonomic neuropathy)
- Elderly patients
- Women
- May present only as dyspnoea, weakness, confusion, or unexplained hypotension
- Dyspnoea
- Epigastric discomfort
- Nausea
- Profound weakness
Physical Signs (Harrison's)
| Finding | Significance |
|---|---|
| Anxiety, restlessness | Patient tries to relieve pain by moving |
| Pallor, diaphoresis, cool peripheries | Sympathetic activation, low CO |
| Tachycardia + hypertension | Anterior MI — sympathetic hyperactivity |
| Bradycardia + hypotension | Inferior MI — parasympathetic hyperactivity (vagal) |
| S4 heart sound | Reduced LV compliance (almost always present in AMI) |
| S3 heart sound | Suggests LV failure |
| Soft S1 | Decreased LV contractility |
| Paradoxical S2 splitting | LBBB or LV dysfunction |
| Apical systolic murmur | Papillary muscle dysfunction → MR |
| Pericardial friction rub | Transmural MI → pericarditis (day 2–4) |
| Raised JVP + clear lungs + hypotension | RV infarction (inferior MI) |
| Fever (up to 38°C) | Non-specific inflammatory response (first week) |
Killip Classification (Severity of LV Failure in AMI)
| Class | Features | Mortality |
|---|---|---|
| I | No signs of heart failure | ~6% |
| II | Mild-moderate HF: S3, basal rales | ~17% |
| III | Severe HF: pulmonary oedema | ~38% |
| IV | Cardiogenic shock: BP < 90, oliguria, peripheral shutdown | ~67% |
PART 4: INVESTIGATIONS
1. Electrocardiogram (ECG) — Most Important Initial Investigation
| Stage | ECG Change |
|---|---|
| Hyperacute (minutes) | Tall, peaked hyperacute T waves |
| Early (hours) | ST elevation ≥ 1 mm in ≥ 2 contiguous leads (tombstone pattern) |
| Hours–days | T wave inversion |
| Days–weeks | Pathological Q waves (> 1 mm wide, > 25% of R wave) |
- ST depression and/or T-wave inversion (no ST elevation, no Q waves)
| Territory | Leads Involved | Artery |
|---|---|---|
| Anterior | V1–V4 | LAD |
| Anterolateral | V1–V6, I, aVL | LAD / LCx |
| Lateral | I, aVL, V5–V6 | LCx |
| Inferior | II, III, aVF | RCA (80%) |
| Posterior | ST depression V1–V3 (reciprocal) | RCA / LCx |
| Right ventricular | V3R–V4R (right-sided leads) | RCA |
2. Cardiac Biomarkers
| Biomarker | Rises | Peaks | Normalises | Notes |
|---|---|---|---|---|
| Myoglobin | 1–2 h | 4–6 h | 24 h | First to rise; poor specificity |
| CK-MB | 2–4 h | 24–48 h | 72 h | Useful for re-infarction |
| Troponin I / T | 2–4 h | 24 h | 7–14 days | Gold standard; highest sensitivity + specificity |
| hs-cTn (high-sensitivity) | < 1 hour | — | — | Allows rapid rule-in/rule-out (0h/1h algorithm) |
Troponin levels rise 20–50 times the upper reference limit (99th percentile) in classic MI. Recanalization causes early peak ("washout" phenomenon) — useful to confirm reperfusion.
- WBC: Rises within hours, peaks 12,000–15,000/μL, persists 3–7 days
- ESR: Rises more slowly, peaks first week, may remain elevated 1–2 weeks
- CRP: Rises with inflammation
3. Cardiac Imaging
- Regional wall motion abnormalities — almost universally present
- LV ejection fraction (EF) — critical for prognosis and therapy decisions
- Detect: RV infarction, LV aneurysm, pericardial effusion, LV thrombus, MR (papillary rupture), VSD
- Doppler: identifies mitral regurgitation, VSD
- Gold standard for identifying the culprit lesion
- Guides primary PCI — simultaneous diagnostic and therapeutic
- Pulmonary oedema (Kerley B lines, bat-wing pattern, pleural effusion)
- Cardiomegaly
- Exclude aortic dissection, pneumothorax
- Radionuclide scanning (SPECT/PET): perfusion defects in ischaemic zones
- MRI: Late gadolinium enhancement → infarct size and viability; not first-line in acute setting
PART 5: MANAGEMENT
A. IMMEDIATE MANAGEMENT (First 30 Minutes — "MONA + Heparin")
Time is myocardium — Goal: Door-to-balloon < 90 minutes (primary PCI)
| Drug | Dose / Details | Rationale |
|---|---|---|
| Morphine | 2–4 mg IV (repeat 5-min intervals) | Analgesia; reduces sympathetic activation; use cautiously (↓ BP, nausea) |
| Oxygen | Only if SpO₂ < 90% | Correct hypoxaemia; not routinely for normoxic patients |
| Nitrates | Sublingual GTN 0.4 mg × 3 doses 5 min apart; then IV if needed | ↓ preload, coronary vasodilation; avoid in hypotension (SBP < 90), RV infarction, phosphodiesterase-5 inhibitor use |
| Aspirin | 160–325 mg chewed immediately | Irreversible COX-1 inhibition → ↓ TxA₂ → antiplatelet |
| P2Y₁₂ inhibitor | Ticagrelor 180 mg OR Clopidogrel 300–600 mg | DAPT with aspirin → prevents reocclusion |
| Anticoagulation | UFH 60 U/kg IV bolus (max 4000 U) + infusion OR Enoxaparin (LMWH) OR Fondaparinux | Prevents thrombus extension; adjunct to reperfusion |
| Beta-blocker | Metoprolol 25–50 mg oral (IV if hypertensive/tachycardic) | ↓ HR, ↓ O₂ demand, ↓ infarct size, antiarrhythmic; avoid in HF, bradycardia, hypotension |
| Statin | Atorvastatin 80 mg or Rosuvastatin 40 mg | Plaque stabilisation, anti-inflammatory; start immediately |
B. REPERFUSION THERAPY (Core of STEMI Management)
- Balloon angioplasty + drug-eluting stent (DES) to culprit artery
- Superior to thrombolysis: higher patency, lower reocclusion, less bleeding
- Goal: Door-to-balloon < 90 minutes
| Agent | Dose |
|---|---|
| Tenecteplase (TNK-tPA) | Single weight-based IV bolus |
| Alteplase (tPA) | Accelerated regimen over 90 min |
| Streptokinase | 1.5 million units over 60 min |
- Previous haemorrhagic stroke (absolute)
- Ischaemic stroke within 3 months
- Active internal bleeding
- Suspected aortic dissection
- Recent major surgery (< 3 weeks)
- Severe uncontrolled hypertension (SBP > 180)
C. PHARMACOTHERAPY (In-Hospital)
| Drug Class | Agent | Indication |
|---|---|---|
| DAPT | Aspirin + Ticagrelor/Clopidogrel | Continue for 12 months post-MI |
| Anticoagulation | Enoxaparin / UFH | During admission |
| ACE Inhibitor | Ramipril / Lisinopril | Start within 24h if EF < 40%, anterior MI, HTN, DM; reduces LV remodelling |
| Beta-blocker | Metoprolol / Carvedilol | Reduce mortality, arrhythmias; start when stable |
| Statin | High-intensity statin | LDL target < 1.8 mmol/L (70 mg/dL) |
| Aldosterone antagonist | Eplerenone / Spironolactone | EF < 35% + HF symptoms or diabetes |
| Nitrates | Sublingual / oral / IV | Symptom control |
D. CCU (Coronary Care Unit) Monitoring
- Continuous cardiac monitoring (arrhythmia detection)
- Haemodynamic monitoring (BP, urine output)
- Bed rest first 6–12 hours; gradual mobilisation by 24 h
- Discharge in 3–5 days if uncomplicated
E. COMPLICATIONS AND THEIR MANAGEMENT
| Complication | Timing | Management |
|---|---|---|
| Ventricular Fibrillation | Immediate | Defibrillation (200–360 J), CPR, Amiodarone |
| Complete Heart Block (inferior MI) | Hours | Atropine; temporary pacing |
| LV Failure / Pulmonary oedema | Hours–days | O₂, diuretics (furosemide), ACE-I |
| Cardiogenic Shock | Hours–days | Inotropes (dobutamine), IABP/Impella; Emergency PCI |
| Free wall rupture | Day 3–7 | Surgical emergency; pericardiocentesis for tamponade |
| VSD | Day 3–7 | Surgical repair; IABP bridge |
| Papillary muscle rupture | Day 3–7 | Surgical mitral valve repair/replacement |
| Pericarditis (early) | Day 2–4 | Aspirin, NSAIDs (avoid steroids) |
| Dressler's syndrome | Weeks–months | NSAIDs, Colchicine |
| Mural thrombus | Days–weeks | Anticoagulation (warfarin/NOAC) for 3–6 months |
| LV Aneurysm | Weeks–months | Persistent ST elevation; anti-failure; surgery if refractory |
F. SECONDARY PREVENTION (Long-Term)
| Intervention | Target/Detail |
|---|---|
| DAPT | Aspirin + P2Y₁₂ for 12 months, then aspirin lifelong |
| Beta-blocker | Lifelong if LV dysfunction |
| ACE inhibitor / ARB | Lifelong if EF < 40% or hypertension |
| High-intensity statin | LDL < 70 mg/dL |
| Aldosterone antagonist | If EF < 35% |
| ICD implantation | If EF < 35% after ≥ 40 days |
| Cardiac rehabilitation | Exercise programme; improves outcomes |
| Lifestyle modification | Smoking cessation, diet, exercise, weight loss |
SUMMARY — HIGH-YIELD EXAM POINTS
| Point | Detail |
|---|---|
| Most common cause | Plaque rupture → thrombosis (Type 1 MI) |
| Most common symptom | Crushing central chest pain > 30 min, not relieved by GTN |
| Most common cause of pre-hospital death | Ventricular fibrillation |
| Gold standard biomarker | Cardiac Troponin I / T |
| Best initial investigation | 12-lead ECG immediately |
| STEMI ECG | ST elevation → T inversion → Q waves |
| NSTEMI ECG | ST depression / T inversion (no Q waves) |
| Reperfusion target | Door-to-balloon < 90 minutes (primary PCI) |
| Immediate drug (first given) | Aspirin 300 mg chewed |
| "Time is myocardium" drug | Primary PCI (preferred over thrombolysis) |
| RV infarction triad | Hypotension + raised JVP + clear lungs (inferior MI) |
| Avoid in RV infarction | Nitrates (drop BP critically) |
| Mechanical complication day 3–7 | Free wall rupture, VSD, papillary muscle rupture |
| Killip IV | Cardiogenic shock — highest mortality (~67%) |
I need to write the pathogenesis
acute myocardial infarction pathogenesis plaque rupture thrombus coronary occlusion diagram
PATHOGENESIS OF ACUTE MYOCARDIAL INFARCTION
Harrison's 22e + Robbins, Cotran & Kumar — Final Year MBBS
DEFINITION
"MI is defined as the presence of acute myocardial injury detected by abnormal cardiac biomarkers in the setting of evidence of acute myocardial ischaemia." — Universal Definition of MI (4th edition, 2018), cited in Harrison's 22e
OVERVIEW — TWO-HIT CONCEPT (Harrison's)
| Vulnerability | Examples |
|---|---|
| Vulnerable vessel | Thin-capped atherosclerotic plaque prone to rupture |
| Vulnerable blood | Hyperglycaemia, elevated LDL, fibrinogen, tissue factor, platelet microparticles → hypercoagulable state |
The overlap of these two promotes hypercoagulability + hypofibrinolysis, especially in diabetics.
STEP-BY-STEP PATHOGENESIS
STEP 1 — Atherosclerotic Plaque (The Substrate)
- Over years, atherosclerosis silently narrows coronary arteries
- A vulnerable (unstable) plaque has:
- Large lipid-rich necrotic core (> 40% of plaque volume)
- Thin fibrous cap (< 65 µm)
- Dense inflammatory infiltrate (macrophages, T-cells at shoulder region)
- Expansive remodelling, neovascularisation, plaque haemorrhage, adventitial inflammation, "spotty" calcification
- Key point: Most plaques causing STEMI did NOT have critical (> 70%) stenosis before rupture — it is plaque composition, not size, that determines rupture risk
"Slowly developing, high-grade coronary artery stenoses do not typically precipitate STEMI because of the development of a rich collateral network over time." — Harrison's 22e
STEP 2 — Plaque Disruption (The Trigger)
| Mechanism | Description |
|---|---|
| Plaque Rupture (65–75%) | Physical tearing of the thin fibrous cap; exposes lipid core + collagen to blood |
| Plaque Erosion (25–35%) | Superficial endothelial loss without full cap rupture; more common in young women, smokers |
- Intrinsic: Macrophages secrete MMPs (Matrix Metalloproteinases) → collagen degradation → cap weakening; T-cell IFN-γ → inhibits SMC collagen synthesis → net thinning
- Extrinsic: Adrenergic surge (morning awakening, exercise, stress) → BP spike → shear forces → cap tears
- Risk factors: cigarette smoking, hypertension, lipid accumulation, systemic inflammation
STEP 3 — Platelet Activation and Aggregation

- Subendothelial collagen + necrotic plaque contents exposed to blood
- Platelets adhere to exposed collagen (via GP Ib–vWF axis) → form initial monolayer
- Platelet activation by agonists: collagen, ADP, epinephrine, serotonin
- Activated platelets release Thromboxane A₂ (TxA₂):
- Potent vasoconstriction → further narrows lumen
- Amplifies further platelet activation
- Creates resistance to fibrinolysis
- GP IIb/IIIa receptor undergoes conformational change → high affinity for fibrinogen → fibrinogen bridges two platelets → platelet cross-linking and aggregation
STEP 4 — Coagulation Cascade Activation → Thrombus Formation
- Tissue factor (expressed in vascular smooth muscle cells and macrophages of the plaque) is exposed at the rupture site
- Tissue factor activates Factor VII → Factor X (extrinsic pathway)
- Prothrombin → Thrombin (via Factor Xa + Va)
- Thrombin → converts Fibrinogen → Fibrin strands
- Thrombin autoamplification — fluid-phase and clot-bound thrombin further accelerate cascade
- Fibrin strands + platelet aggregates + trapped RBCs → occlusive coronary thrombus forms within minutes
"The culprit coronary artery eventually becomes occluded by a thrombus containing platelet aggregates and fibrin strands." — Harrison's 22e
- Complete occlusion → STEMI (transmural infarction)
- Partial/stuttering occlusion → NSTEMI / Unstable Angina (subendocardial infarction)
STEP 5 — Myocardial Response to Ischaemia
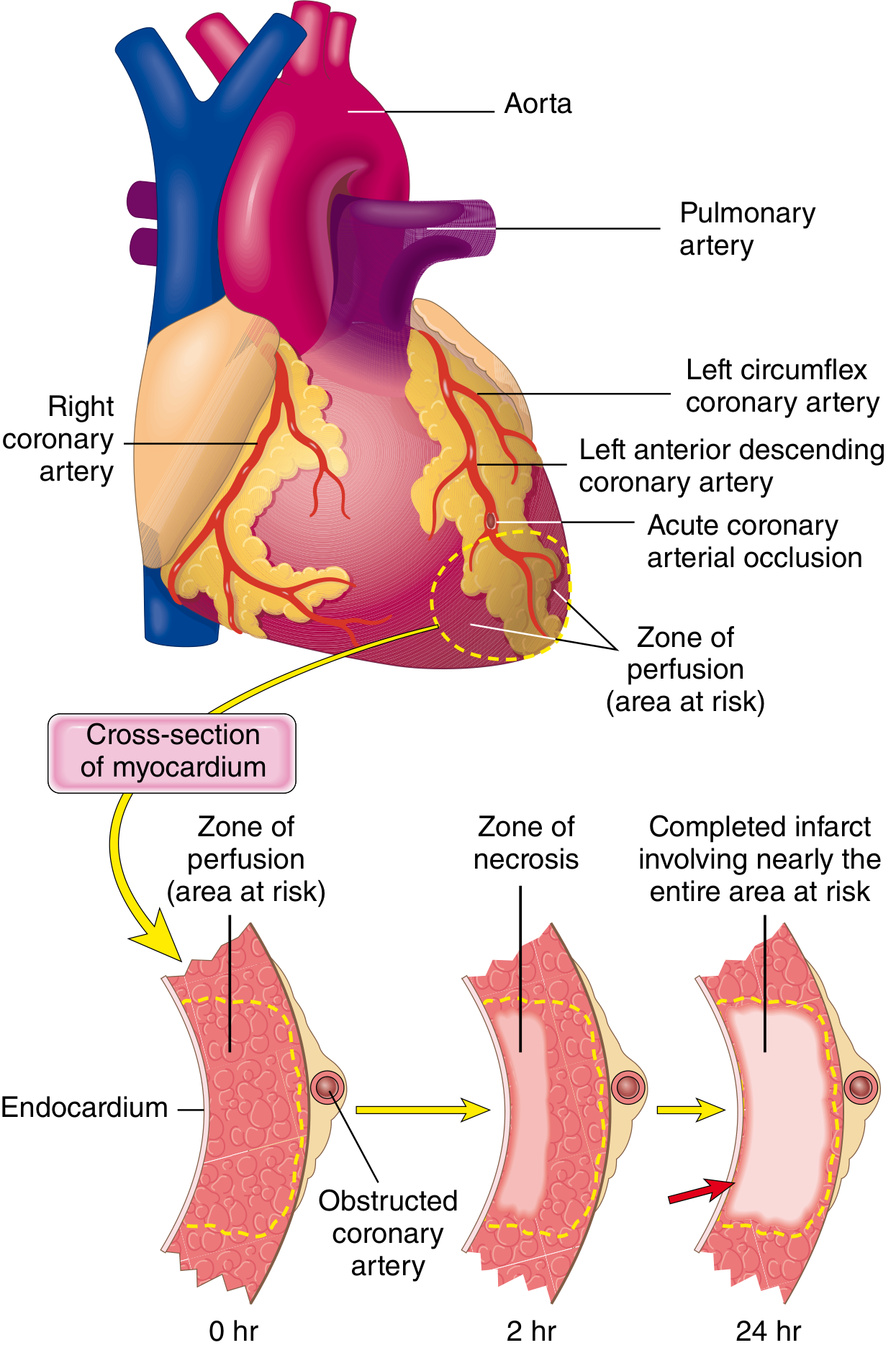
| Time | Event |
|---|---|
| Seconds | Aerobic metabolism ceases → ATP depletion begins; lactic acid accumulates |
| < 2 minutes | Loss of myocardial contractility |
| 10 minutes | ATP reduced to 50% of normal |
| 20–40 minutes | Irreversible cell injury (necrosis) begins ← Key exam point |
| 40 minutes | ATP reduced to 10% of normal |
| > 1 hour | Microvascular injury follows myocyte death |
| 6–12 hours | Progressive necrosis complete throughout area at risk |
- ↓ ATP → failure of Na⁺/K⁺ ATPase → cell swelling
- Lactic acid accumulation → intracellular acidosis → enzyme dysfunction
- ↑ Intracellular Ca²⁺ → mitochondrial dysfunction → cell death
- Sarcolemmal rupture → intracellular proteins (Troponin, CK-MB, myoglobin) leak into circulation → forms the basis of cardiac biomarkers
- Necrosis begins in the subendocardium first (most vulnerable — furthest from epicardial supply + highest wall stress)
- Progresses outward like a wavefront toward the epicardium with prolonged ischaemia
- Reperfusion within 20–40 min can abort this wavefront and salvage myocardium
STEP 6 — Reperfusion and Its Consequences
- Restores flow → salvages myocardium still alive at margins of infarct
- Limits infarct size; improves LV function and survival
- Despite being net beneficial, reperfusion can cause:
- Contraction band necrosis (hypercontraction of myofibrils — hallmark microscopically)
- Free radical damage (reactive oxygen species burst on reperfusion)
- Stunning — post-ischaemic myocardium that is alive but dysfunctional for days after reperfusion
STEP 7 — Causes of MI Without Atherothrombosis (~10% of cases)
| Mechanism | Example |
|---|---|
| Coronary vasospasm | Cocaine, Prinzmetal angina |
| Embolism | AF (left atrial thrombus), infective endocarditis, prosthetic valves |
| Increased demand / supply mismatch (Type 2 MI) | Severe tachycardia, anaemia, shock |
| Vasculitis | Small intramyocardial vessel inflammation |
| Aortic dissection | Retrograde dissection into coronary ostium |
| SCAD | Spontaneous coronary artery dissection (young women) |
| Sickle cell disease | Vascular stasis and thrombosis |
EVIDENCE SUPPORTING THROMBOTIC PATHOGENESIS
| Evidence | Finding |
|---|---|
| Angiography within 4 hours of MI | Coronary thrombosis in ~90% of patients |
| Angiography at 12–24 hours (no treatment) | Thrombosis found in only 60% — some clear spontaneously |
| Thrombolysis + PCI within golden hour | Successfully restores patency; limits infarct size |
| Autopsy studies | Plaque disruption + overlying thrombus at culprit site |
SUMMARY FLOWCHART — EXAM READY
RISK FACTORS (HTN, DM, Smoking, Hyperlipidaemia, Age, Family Hx)
↓
VULNERABLE ATHEROSCLEROTIC PLAQUE
(thin cap + large lipid core + inflammation)
↓
PLAQUE RUPTURE or EROSION
(MMPs degrade cap; adrenergic surge triggers mechanical disruption)
↓
Subendothelial collagen + plaque contents exposed to blood
↓
PLATELET ADHESION → ACTIVATION → TxA₂ released
↓
GP IIb/IIIa activation → fibrinogen binding → PLATELET AGGREGATION
↓
TISSUE FACTOR exposed → Extrinsic pathway activated
↓
Prothrombin → Thrombin → Fibrinogen → FIBRIN
↓
OCCLUSIVE CORONARY THROMBUS
↓
Complete → STEMI Partial → NSTEMI / UA
↓
CESSATION OF BLOOD FLOW → Area at risk
↓
Seconds: ATP depletion, lactic acid ↑
< 2 min: Loss of contractility
20–40 min: IRREVERSIBLE NECROSIS begins
↓
SUBENDOCARDIAL NECROSIS → WAVEFRONT → TRANSMURAL INFARCTION (6–12 hr)
↓
Sarcolemmal rupture → TROPONIN/CK-MB leak into blood (diagnostic biomarkers)
HIGH-YIELD EXAM POINTS
| Point | Fact |
|---|---|
| Underlying cause | Atherosclerotic plaque rupture + coronary thrombosis (90%+ of cases) |
| Vulnerable plaque features | Thin cap + large lipid core + macrophage inflammation |
| Role of MMPs | Macrophage-derived; degrade fibrous cap collagen → rupture |
| Platelet key receptor | GP IIb/IIIa — cross-links platelets via fibrinogen |
| Vasospasm mediator | Thromboxane A₂ (also: serotonin, ADP) |
| Coagulation trigger | Tissue factor → Factors VII + X → thrombin → fibrin |
| Point of NO RETURN | 20–40 minutes of severe ischaemia |
| First area to die | Subendocardium (wavefront phenomenon) |
| Biomarker basis | Sarcolemmal rupture → intracellular proteins leak into blood |
| Most sensitive/specific biomarker | Cardiac Troponin I / T |
| Reperfusion hallmark histology | Contraction band necrosis |
| Reperfusion benefit | Salvages myocardium if done within 20–40 min of ischaemia |
This etiopathogenesis will be for maximum 3 marks . Won’t be able to write such long
ETIOPATHOGENESIS OF ACUTE MI
(3-mark answer — ~10–12 lines)
ETIOLOGY
- Coronary vasospasm (cocaine, Prinzmetal angina)
- Coronary embolism (AF, infective endocarditis)
- Type 2 MI — supply-demand mismatch (severe anaemia, tachycardia, shock)
PATHOGENESIS
Key exam lines:
- Most MI caused by plaque rupture + thrombosis, not by critical stenosis
- Irreversible necrosis begins at 20–40 minutes
- GP IIb/IIIa + Tissue factor are the key molecular triggers
Bhai ek baar Hindi mein Bata de bas Jo Abhi bola
Bhai ek baar Hindi mein Bata de bas Jo Abhi bola