Mechanism of gdm
Reading File
Finding Sources
Searching PubMed
"gestational diabetes" AND "insulin resistance" AND mechanism
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Reading File
Loading Image
Finding Sources
I now have comprehensive content from authoritative textbooks. Here is the full answer:
Mechanism of Gestational Diabetes Mellitus (GDM)
GDM is defined as glucose intolerance first recognized during the second or third trimester of pregnancy. The core pathophysiology is a two-hit model: progressive pregnancy-induced insulin resistance (hit 1) that outstrips the compensatory capacity of the maternal beta cells (hit 2).
1. Normal Metabolic Adaptations of Pregnancy
In early pregnancy, metabolic changes favor anabolism - insulin sensitivity is actually slightly increased. By the second trimester, the metabolic environment shifts dramatically toward insulin resistance. This is physiologically purposeful: making maternal glucose less available to maternal tissues and redirecting it to the fetus via facilitated diffusion across the placenta.
Key basal changes by late pregnancy (from Creasy & Resnik's Maternal-Fetal Medicine):
| Parameter | Non-Pregnant | Late Pregnancy |
|---|---|---|
| Fasting glucose | 79 ± 2.4 mg/dL | 68 ± 1.5 mg/dL (lower) |
| Fasting insulin | 9.8 ± 1.1 µU/mL | 16.2 ± 2.0 µU/mL (higher) |
| Plasma amino acids | 3.82 µmol/L | 3.18 µmol/L (lower - shunted to fetus) |
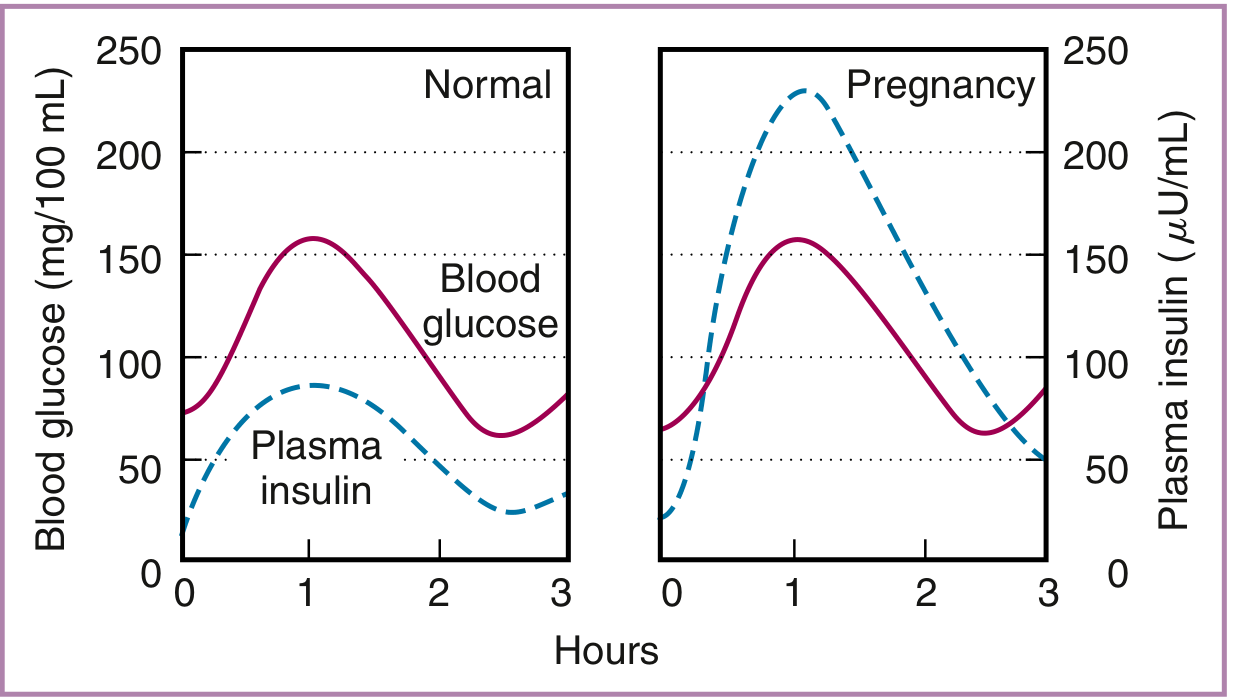
The graph below shows the dramatic compensatory hyperinsulinemia in late pregnancy - plasma insulin rises to ~230 µU/mL after an oral glucose load, compared to ~80 µU/mL in non-pregnant women, yet blood glucose levels follow a similar curve:
2. The Diabetogenic Hormones of the Placenta
The primary drivers of insulin resistance are placental hormones that increase progressively from the second trimester onward. The rapid normalization of glucose metabolism after delivery is the best evidence that these hormones are the major diabetogenic factors.
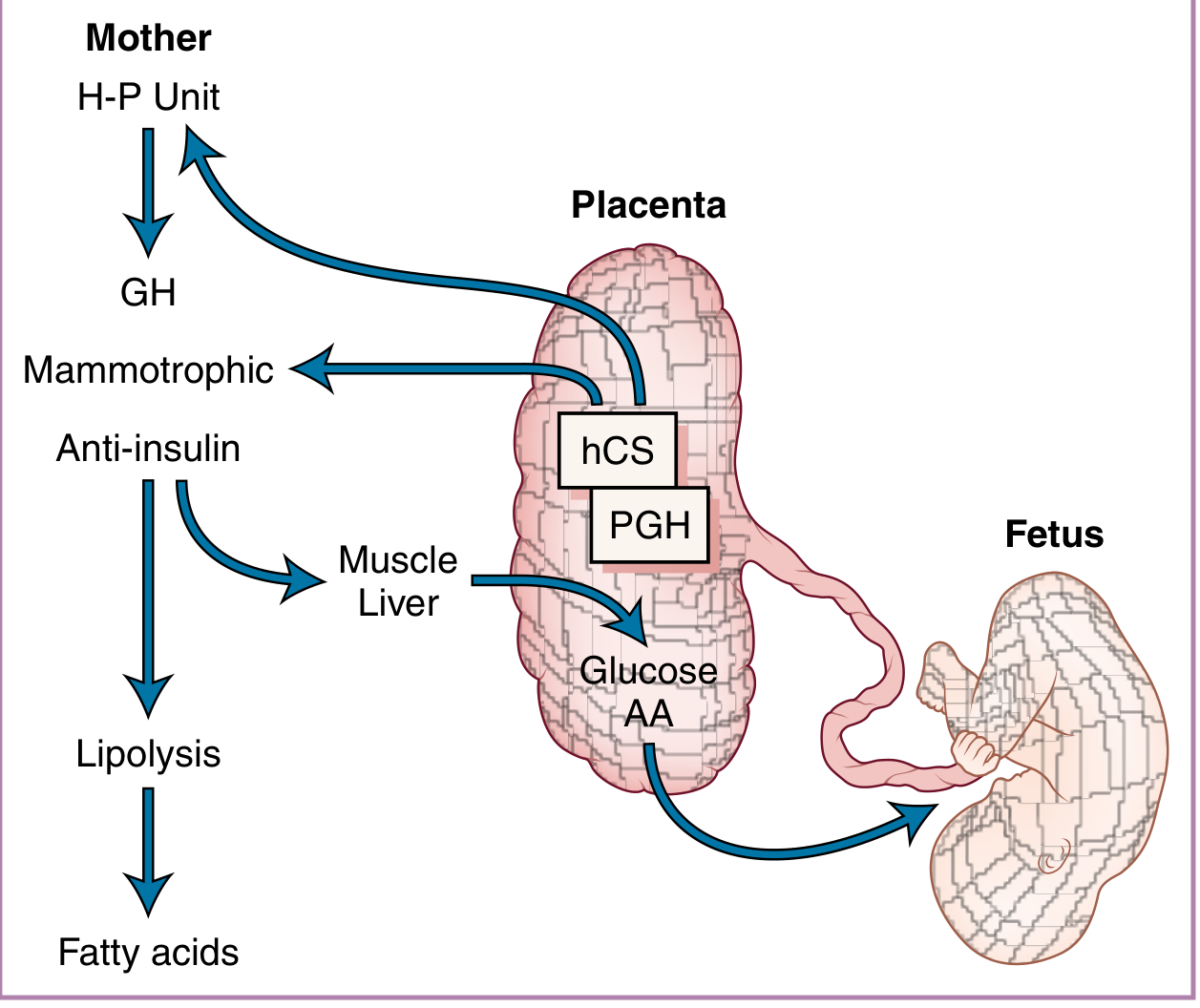
Human Chorionic Somatomammotropin (hCS) / Human Placental Lactogen (hPL)
- Structurally similar to GH and prolactin
- Reduces insulin receptor sites on insulin-sensitive tissues (muscle, liver, adipose)
- Impairs post-receptor glucose transport (downregulates GLUT4 translocation)
- Promotes lipolysis → raises free fatty acids (FFAs) → FFAs directly inhibit insulin signaling (Randle cycle effect)
- Levels rise proportionally with placental mass
Human Placental Growth Hormone (hPGH)
- Encoded by hGH-V gene; secreted in a non-pulsatile pattern (unlike pituitary GH)
- Replaces pituitary GH in maternal circulation by mid-pregnancy
- Stimulates IGF-1 production, which feeds back to suppress pituitary GH
- Overexpression in mice causes severe insulin resistance with elevated fasting insulin and impaired glucose tolerance - demonstrating its central role
Other counter-regulatory hormones that amplify resistance:
- Progesterone - inhibits insulin-stimulated glucose uptake; reduces beta-cell secretory response
- Cortisol - increases hepatic gluconeogenesis; promotes peripheral insulin resistance
- Prolactin - has complex effects but contributes to altered insulin sensitivity
The diagram above shows the proposed functional roles of hCS and PGH: they create an anti-insulin, lipolytic state in the mother that diverts glucose and amino acids (AA) preferentially to the fetus.
3. Beta-Cell Compensation - and Why It Fails in GDM
In a normal pregnancy, the pancreatic islets mount a robust compensatory response:
- Beta-cell hyperplasia (islet mass increases)
- Augmented first- and second-phase insulin secretion
- Higher peak insulin levels after glucose loads
This is why most pregnant women do not develop GDM despite having significant insulin resistance.
Women who develop GDM have a pre-existing, subclinical beta-cell defect - often the same defect that predisposes them to type 2 DM later in life. Their islets cannot secrete enough insulin to overcome the placental hormone-driven resistance. The result is:
- Impaired first-phase insulin secretion (blunted immediate response to glucose)
- Relative hyperglycemia, especially postprandial
- Glucose intolerance meeting criteria for GDM
This is why ~50% of women with GDM go on to develop T2DM within 10 years - the underlying beta-cell insufficiency persists after the placental hormones are removed.
4. Additional Molecular Contributors
- Insulin receptor signaling defects - Reduced IRS-1 and PI3K activity in skeletal muscle, impairing GLUT4 translocation to the cell surface
- Inflammatory cytokines - TNF-alpha (produced by placenta and adipose tissue) interferes with insulin receptor substrate phosphorylation; levels correlate with degree of insulin resistance
- Adipokines - Altered adiponectin (reduced) and leptin (elevated) levels in obese/GDM pregnancies further impair insulin sensitivity
- Free fatty acids - hCS-driven lipolysis raises FFAs, which activate serine kinases that phosphorylate IRS-1 (inhibitory phosphorylation), impairing insulin signaling
5. Summary Flowchart
Pregnancy (2nd-3rd trimester)
↓
Placenta secretes hCS, hPGH, progesterone, cortisol, prolactin
↓
↓ Insulin receptor sites + ↓ GLUT4 translocation + ↑ FFAs
↓
Progressive peripheral insulin resistance
↓
├─── Normal beta cells: hyperplasia + ↑↑ insulin secretion → euglycemia
│
└─── Defective beta cells (GDM): inadequate compensation
↓
Postprandial hyperglycemia → GDM
Key Clinical Correlates
- GDM typically presents in the 2nd/3rd trimester (when placental hormone levels peak), not the 1st
- Resolves postpartum in most women as placental hormones are cleared within hours-days
- 35-60% risk of developing T2DM within 10-20 years - the underlying beta-cell defect remains
- Children born to GDM mothers have increased risk of metabolic syndrome and T2DM (fetal programming via intrauterine hyperglycemia)
- According to Harrison's 22nd edition, the IDF estimated 16% of pregnancies worldwide are affected by GDM or preexisting DM (2021 data)
Recent evidence note: A 2024 systematic review (PMID 39519193) on the cellular and molecular pathophysiology of GDM confirms and expands on the role of inflammatory pathways, mitochondrial dysfunction, and epigenetic changes in GDM - consistent with the textbook mechanisms above.
Sources: Creasy & Resnik's Maternal-Fetal Medicine (Principles and Practice), Harrison's Principles of Internal Medicine 22E, Robbins & Cotran Pathologic Basis of Disease
This is a shared conversation. Sign in to Orris to start your own chat.