Gout in biochemistry
Reading File
Finding Sources
Finding Sources
Searching PubMed
gout hyperuricemia pathophysiology
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
I now have everything needed for a comprehensive biochemistry-focused answer on gout. Here is the full response:
Gout - Biochemistry
Gout is a disorder of purine catabolism resulting in hyperuricemia and deposition of monosodium urate (MSU) crystals in joints and soft tissues, producing recurrent attacks of acute inflammatory arthritis.
1. Uric Acid - The End Product of Purine Catabolism
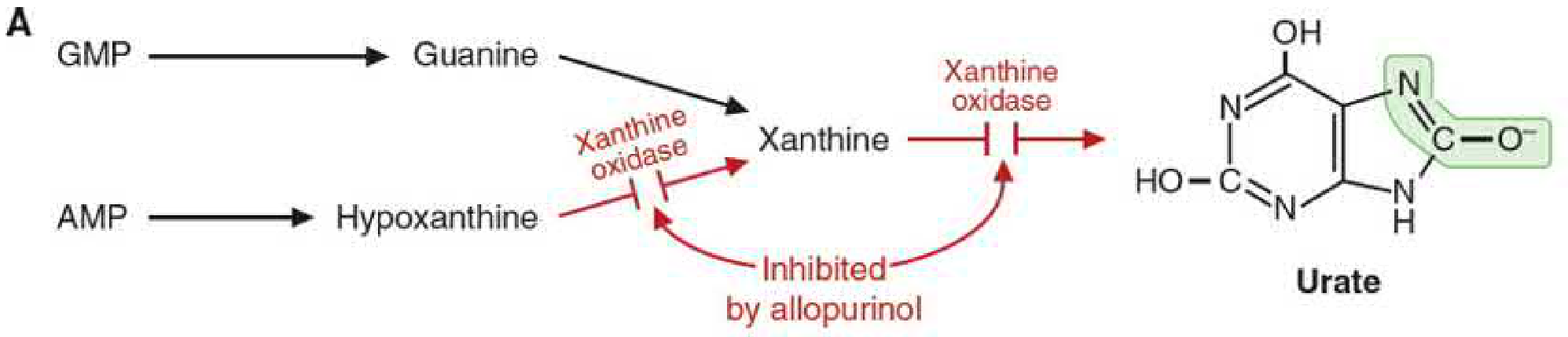
Uric acid is the final, irreversible end product of purine degradation in humans. Unlike most mammals, humans lack the enzyme uricase (urate oxidase), which would otherwise convert uric acid to the more soluble allantoin. This makes uric acid relatively insoluble and prone to crystallization.
Degradation pathway:
- AMP → Adenosine → Inosine → Hypoxanthine
- GMP → Guanosine → Guanine → Xanthine
- Both Hypoxanthine and Xanthine → Uric acid (via xanthine oxidase)
Pyrimidine catabolism does NOT produce uric acid - this is a purely purine phenomenon.
2. Purine Pathways Relevant to Gout
De Novo Synthesis
Purine nucleotides are built from scratch from non-purine precursors (CO₂, glycine, glutamine, aspartate, formyl groups). The first committed step is PRPP + glutamine → phosphoribosylamine, catalyzed by glutamine phosphoribosylpyrophosphate amidotransferase (GPAT), regulated by feedback inhibition from AMP, GMP, and IMP.
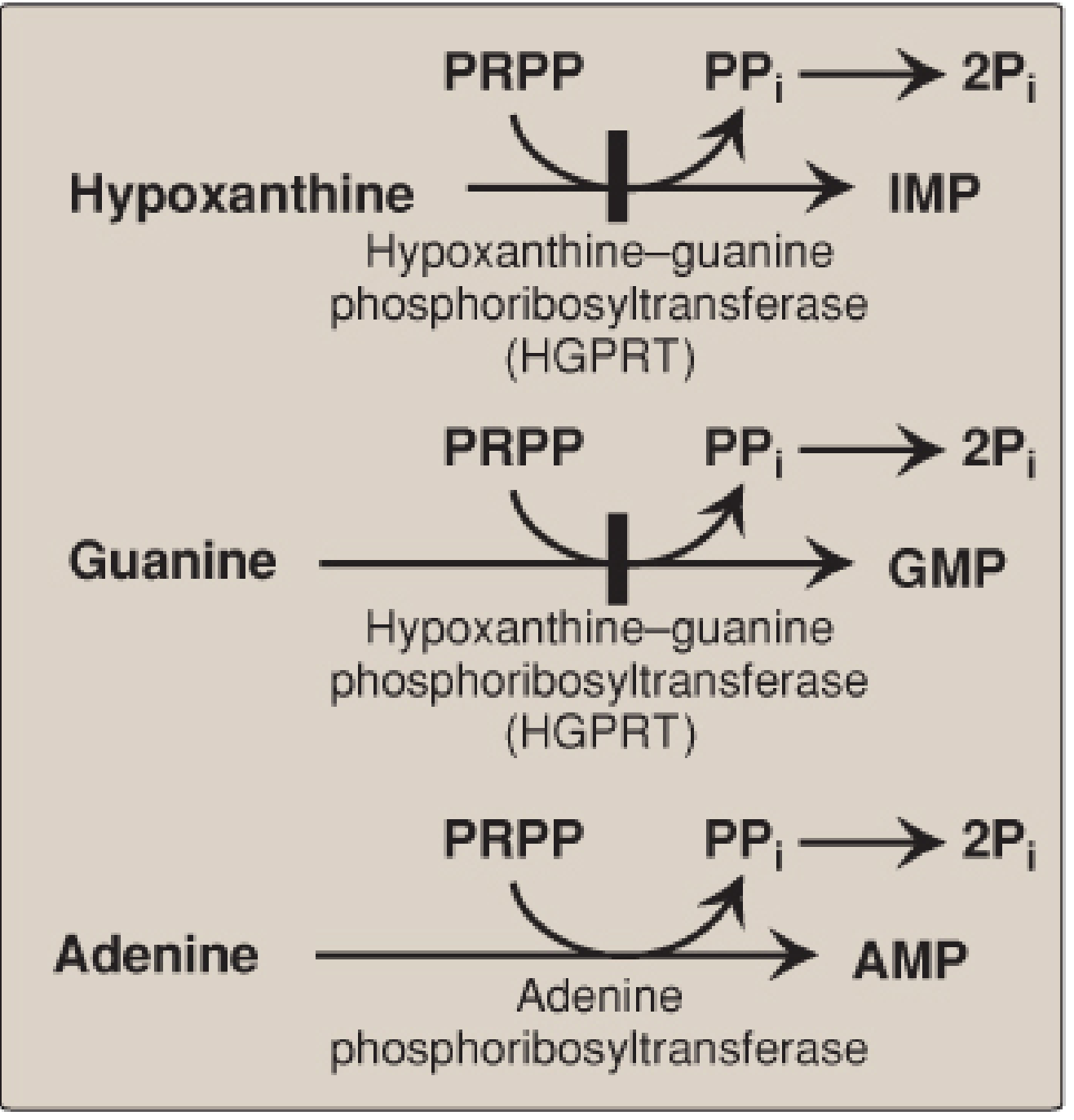
Salvage Pathway (Key to Gout)
Free purine bases from diet or nucleic acid catabolism are recycled back to nucleotides rather than degraded. This is far more energy-efficient than de novo synthesis.
The central enzyme is HGPRT (Hypoxanthine-Guanine Phosphoribosyltransferase):
- Hypoxanthine + PRPP → IMP
- Guanine + PRPP → GMP
A separate enzyme, adenine phosphoribosyltransferase (APRT), salvages adenine → AMP.
3. Causes of Hyperuricemia
Hyperuricemia is defined as plasma urate >6.8 mg/dL (the solubility limit). It results from overproduction, underexcretion, or both.
| Category | Uric Acid Synthesis | Uric Acid Excretion |
|---|---|---|
| Primary gout (unknown enzyme defects) | Normal or ↑ | ↓ or Normal |
| Primary gout (partial HGPRT deficiency) | ↑ | Normal |
| Increased nucleic acid turnover (e.g., leukemia) | ↑↑ | ↑ |
| Chronic renal disease | Normal | ↓ |
| Lesch-Nyhan syndrome (complete HGPRT deficiency) | ↑↑ | ↑ |
Based on Robbins & Cotran Pathologic Basis of Disease, Table 26.6
Most primary gout (>90% of cases) results from reduced renal excretion, not overproduction. Genome-wide studies implicate polymorphisms in transporter genes including URAT1, GLUT9, and KCNQ1.
Secondary causes of overproduction:
- Rapid tumor cell lysis (tumor lysis syndrome during chemotherapy)
- Myeloproliferative disorders
- Lesch-Nyhan syndrome (HGPRT deficiency - see below)
- Increased PRPP synthetase activity
4. Enzymatic Defects Causing Gout
Partial HGPRT Deficiency (Gout without Neurological Features)
When HGPRT activity is partially reduced:
- The salvage pathway is interrupted
- Hypoxanthine and guanine cannot be recycled → degraded to uric acid instead
- PRPP levels rise (less consumed by salvage), stimulating de novo synthesis further
- Net result: hyperuricemia and gout
Lesch-Nyhan Syndrome (Complete HGPRT Deficiency)
- X-linked recessive disorder
- Complete absence of HGPRT
- Massive hyperuricemia + urolithiasis + gouty arthritis
- Plus severe neurological features: intellectual disability, choreoathetosis, spasticity, and compulsive self-mutilation (biting of lips and fingers)
- Classified as a form of secondary gout
As functional HGPRT increases, severity of symptoms decreases - a dose-response relationship.
5. Crystal Deposition and Inflammation
Once plasma urate exceeds the solubility threshold, monosodium urate crystals precipitate in joints (particularly the first metatarsophalangeal joint - "podagra"), bursae, and soft tissues (tophi).
Inflammatory cascade:
- MSU crystals are phagocytosed by synovial macrophages and neutrophils
- Crystals activate the NLRP3 inflammasome (a cytosolic pattern recognition platform)
- Inflammasome activates caspase-1, which cleaves pro-IL-1β → active IL-1β
- IL-1β promotes massive neutrophil recruitment into the joint
- Neutrophils release more cytokines, reactive oxygen species, and proteases
- Crystals can rupture phagolysosomes, releasing lysosomal enzymes
- Result: acute synovitis that typically self-resolves in days to weeks
Repeated attacks eventually lead to tophi (aggregates of urate crystals surrounded by inflammatory cells) and permanent joint damage.
6. Biochemical Basis of Treatment
Xanthine Oxidase Inhibitors (reduce uric acid production)
Allopurinol - the classic example of a suicide (mechanism-based) inhibitor:
- Allopurinol is a purine analog (isomer of hypoxanthine)
- Xanthine oxidase oxidizes allopurinol to oxypurinol (alloxanthine)
- Oxypurinol binds tightly within the molybdenum-sulfide coordination sphere of the active site
- The enzyme has "committed suicide" - permanently inactivated and unable to generate uric acid
- Both conversions (hypoxanthine→xanthine and xanthine→uric acid) are blocked
Febuxostat - a non-purine xanthine oxidase inhibitor; also used for hyperuricemia/gout prevention. A 2024 systematic review (PMID: 38738384) found febuxostat provides additional renoprotective benefits in patients with hyperuricemia or gout.
Uricosuric Agents (increase uric acid excretion)
Probenecid - blocks renal tubular reabsorption of uric acid, increasing urinary excretion. Useful when underexcretion is the primary mechanism.
IL-1 Inhibitors (block inflammatory cascade)
Anakinra, canakinumab - block IL-1β to suppress the inflammasome-driven acute attack. Used in refractory cases.
Colchicine (acute attack)
Disrupts microtubule polymerization, impairing neutrophil migration into the joint - does not lower uric acid.
NSAIDs / Corticosteroids
Anti-inflammatory agents for acute attack management.
7. Other Biochemical Associations
- Glucose-6-phosphatase deficiency (von Gierke disease): Glucose-6-phosphate is diverted to the pentose phosphate pathway, generating excess PRPP, driving de novo purine synthesis and uric acid production.
- Fructose ingestion: Fructose-1-phosphate consumes ATP, leading to AMP accumulation and increased purine degradation.
- Alcohol: Promotes nucleotide turnover and lactate production (lactate competes with urate for renal secretion).
- Diuretics (thiazides, loop): Reduce renal urate excretion, precipitating gout.
Key Summary Points
| Fact | Detail |
|---|---|
| End product of purine catabolism | Uric acid (humans lack uricase) |
| Key degradative enzyme | Xanthine oxidase |
| Key salvage enzyme | HGPRT |
| HGPRT deficiency (partial) | Gout (overproduction) |
| HGPRT deficiency (complete) | Lesch-Nyhan syndrome |
| Crystal type | Monosodium urate (needle-shaped, negatively birefringent) |
| Inflammasome involved | NLRP3 → caspase-1 → IL-1β |
| Allopurinol mechanism | Suicide inhibitor of xanthine oxidase |
| Most common cause of primary gout | Reduced renal excretion |
Sources: Robbins & Kumar Basic Pathology; Robbins, Cotran & Kumar Pathologic Basis of Disease; Lippincott Illustrated Reviews: Biochemistry, 8th ed.; Basic Medical Biochemistry - A Clinical Approach, 6e
This is a shared conversation. Sign in to Orris to start your own chat.