Encephalopathy
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
I now have comprehensive information from multiple authoritative textbooks. Let me compile a thorough response.
Encephalopathy
Encephalopathy is a broad term for diffuse brain dysfunction — any condition causing global impairment of cerebral function, ranging from subtle cognitive changes to deep coma. Unlike focal neurological lesions, encephalopathies typically cause widespread, symmetric disturbances in consciousness, cognition, and behavior.
Classification by Etiology
1. Hepatic Encephalopathy (HE)
Definition: A reversible neuropsychiatric syndrome occurring in patients with chronic liver disease and portal hypertension, or acute liver failure (ALF). It develops in 50–70% of patients with cirrhosis, with 1- and 3-year survival rates of only 42% and 23% without liver transplantation.
Types (by underlying disease):
- Type A — acute liver failure
- Type B — portosystemic bypass/shunts without intrinsic liver disease
- Type C — chronic liver disease with portal hypertension (most common)
Pathophysiology:
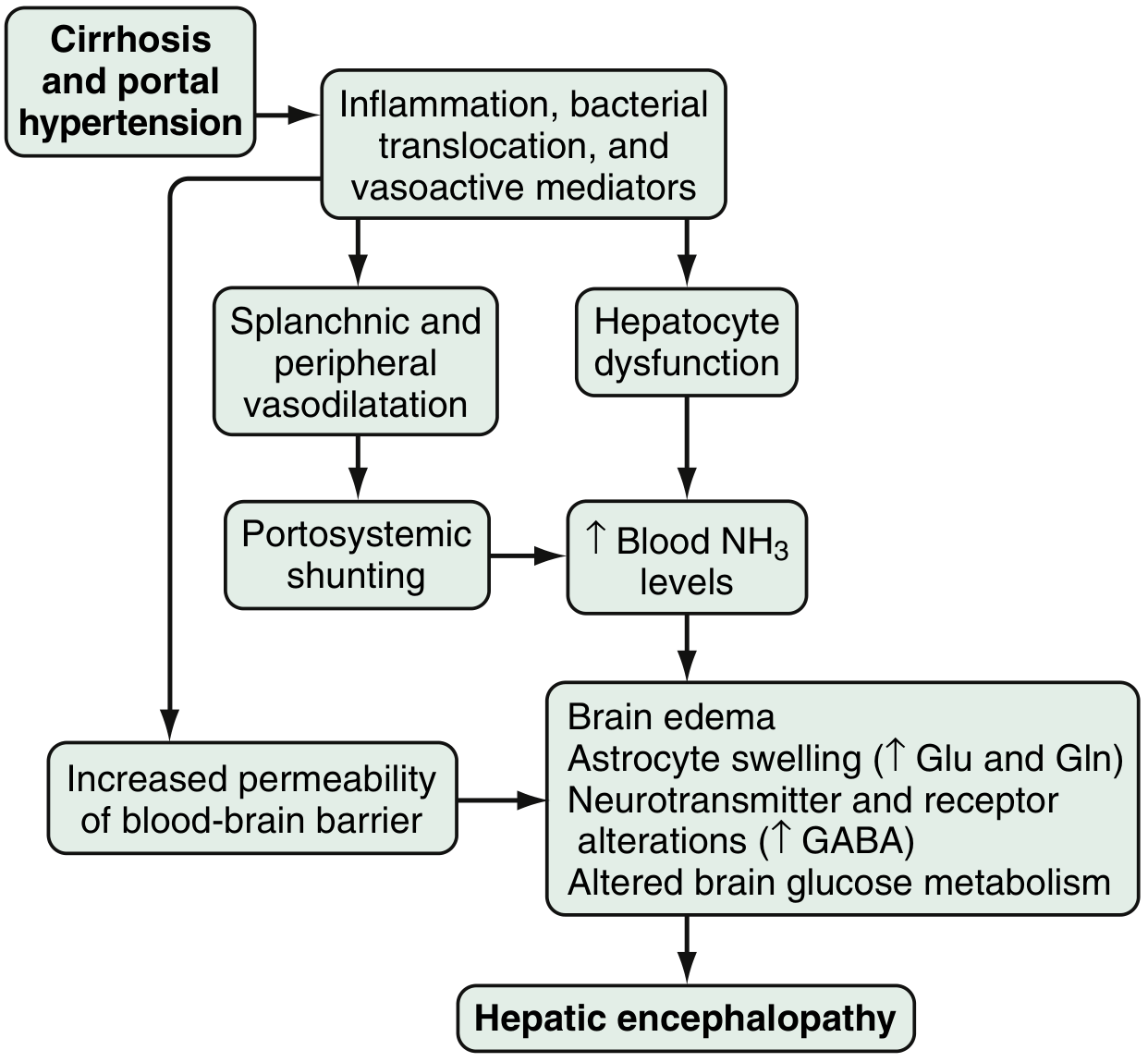
Fig. 94.1 — Proposed pathophysiology of hepatic encephalopathy. — Sleisenger and Fordtran's GI and Liver Disease
The key mechanisms involve:
- Hyperammonemia: Gut bacteria metabolize nitrogenous products to ammonia → enters portal circulation → impaired hepatocyte clearance + portosystemic shunting → ↑ blood NH₃ → astrocyte swelling, brain edema
- GABA-benzodiazepine system: Enhanced astrocyte (peripheral-type) benzodiazepine receptor sensitivity → ↑ GABA-A activation, neuroinhibition; neurosteroids (allopregnanolone) further amplify this
- BBB disruption: Increased permeability → ↑ cerebral uptake of neurotoxins; acute hyperammonemia impairs myoinositol transport
- Neurotransmitter alterations: Serotonin, nitric oxide, circulating opioids, manganese, and oxidative stress all contribute
- Microbiome: Distinct colonic microbiota differences in cirrhotic patients with vs. without HE
Grading (West Haven Criteria / SONIC Classification):
| Grade | Intellectual Function | Neuromuscular | SONIC |
|---|---|---|---|
| 0 | Normal | Normal | Unimpaired |
| Minimal | Subtle work/driving changes | Minor psychometric abnormalities | Covert HE |
| 1 | Shortened attention, altered sleep | Mild asterixis | Covert HE |
| 2 | Disoriented (to time); personality changes | Asterixis, slurred speech | Overt HE |
| 3 | Gross disorientation, bizarre behavior | Asterixis, muscle rigidity | Overt HE |
| 4 | Coma | Decerebrate posturing | Overt HE |
Clinical features: Forgetfulness, handwriting changes, reversed sleep-wake cycle, asterixis ("flapping tremor"), agitation, disinhibition, seizures → coma. Respiratory alkalosis (hyperventilation, low PCO₂, high pH) is a hallmark of severe hepatic coma. Pupillary and oculocephalic reflexes remain normal until the preterminal stage — helping distinguish HE from structural brainstem disease.
Common precipitants: GI bleeding, electrolyte abnormalities, infections, sedative medications, dehydration, high protein intake, constipation.
Treatment:
- Identify and eliminate precipitating factors
- Lactulose 15–45 mL PO bid–qid (first-line) — target 3–5 soft stools/day; can be given as enema (300 mL lactulose + 700 mL water)
- Rifaximin 550 mg PO bid — nonsystemic broad-spectrum antibiotic; reduces recurrence risk significantly in placebo-controlled trials; used alongside lactulose
- Liver transplantation — generally reverses HE
2. Uremic Encephalopathy
Occurs in severe renal failure when uremic toxins accumulate. Symptoms range from early fatigue, drowsiness, restlessness, reduced attention span (often fluctuating) to agitated confusion, psychosis, seizures, and coma. Advanced grades are now mostly seen when dialysis is withheld. Motor signs include action tremor, asterixis, myoclonus, hyperreflexia, and occasionally choreiform movements.
Diagnosis: Clinical picture + severe renal dysfunction, after excluding other causes. EEG shows generalized slowing (↑ theta/delta), bilateral spike-wave complexes; normalizes with therapy. CSF may show ↑ protein (<1 g/L) and mild pleocytosis. Brain imaging is nonspecific (shows only brain volume reduction), unlike hepatic encephalopathy.
Treatment: Renal replacement therapy (dialysis or transplantation) — symptoms typically resolve within days to weeks. — Bradley and Daroff's Neurology in Clinical Practice
3. Wernicke's Encephalopathy
An acute neuropsychiatric emergency caused by thiamine (vitamin B₁) deficiency, most commonly in alcohol dependence (reduced intake + reduced absorption), but also in malnutrition, hyperemesis, dialysis, cancer, HIV/AIDS, or gastric surgery.
Classic triad (present in only ~1/3 of patients):
- Ophthalmoplegia (lateral gaze palsy, nystagmus)
- Ataxia
- Global confusion
Thiamine is required to utilize glucose — a glucose load in a thiamine-deficient patient can precipitate Wernicke's encephalopathy.
Risk factors in alcohol dependence: acute withdrawal, malnourishment, decompensated liver disease, homelessness, peripheral neuropathy, previous WE.
Treatment:
- IV/IM thiamine 200–300 mg once daily for 3–5 days (parenteral first; oral absorption is unreliable)
- If untreated → Korsakoff syndrome (irreversible amnestic disorder) — responds much more slowly and incompletely to thiamine than the acute encephalopathy
4. Sepsis-Associated Encephalopathy (SAE)
The most common form of encephalopathy in the ICU. Presents as diffuse brain dysfunction without prominent focal findings: confusion, disorientation, agitation, fluctuating alertness → coma in severe cases. Hyperreflexia, frontal release signs (grasp, snout reflex), myoclonus, tremor, and asterixis may occur.
Occurs in the majority of patients with sepsis and multisystem organ failure. Mortality of SAE severe enough to cause coma approaches 50%, reflecting severity of the underlying illness rather than brain injury per se. Biomarkers: elevated serum S-100β and neuropathologic findings of neuronal apoptosis and cerebral ischemic injury in fatal cases.
Long-term: Cognitive dysfunction resembling dementia is increasingly recognized in survivors, especially older patients.
Treatment: Address the underlying infection/critical illness — encephalopathy almost always improves substantially with successful treatment. — Harrison's Principles of Internal Medicine 22E
5. Hypertensive Encephalopathy / PRES
Posterior Reversible Encephalopathy Syndrome (PRES) is characterized by headache, altered mental status, visual disturbance, and seizures (seizures in up to 90% of cases). Diagnosis is radiographic — vasogenic edema predominantly in posterior white matter on MRI.
Common associations: hypertensive crisis, eclampsia, vasculitis, thrombotic microangiopathy, calcineurin inhibitors (cyclosporine, tacrolimus), rituximab, sirolimus, erythropoietin. End-stage kidney disease (ESKD) on dialysis is a recognized setting.
Pathogenesis: failure of cerebral autoregulation + endothelial dysfunction → forced vasodilation → vasogenic edema. Clinical manifestations are usually reversible with BP control and volume management.
6. Toxic Encephalopathy
Both bacterial pathogens and the inflammatory response they generate cause neuronal injury via: reactive oxygen species, proteases, cytokines, and excitatory amino acids — leading to both apoptosis and necrosis. Bacterial meningitis frequently causes a toxic encephalopathy with drowsiness/stupor accompanied by fever, tachycardia, and tachypnea (classic triad of fever + nuchal rigidity + AMS present in only 44% of community-acquired meningitis cases).
7. Radiation Encephalopathy
Classified by time of onset after irradiation:
| Phase | Timing | Features | Course |
|---|---|---|---|
| Acute | Days | Headache, nausea, altered mental status; requires >3 Gy to large brain volume | Responds to corticosteroids |
| Early delayed | 2 weeks–4 months | Demyelination; headache, drowsiness, enhanced focal deficits | Self-resolving over weeks |
| Late delayed | Months–years (>35 Gy) | Cognitive/affective disturbances, gait disorder, dementia; necrosis/atrophy | Progressive; bevacizumab may help |
General Diagnostic Approach
| Investigation | Relevance |
|---|---|
| Serum ammonia | HE (present in ~90%; but neither sensitive nor specific alone) |
| LFTs, coagulation | Hepatic cause |
| BUN/creatinine | Uremic cause |
| Blood glucose | Hypoglycemic encephalopathy |
| Electrolytes (Na⁺, Ca²⁺) | Hyponatremia, hypercalcemia |
| Thyroid function | Hypo/hyperthyroid encephalopathy |
| Thiamine level | Wernicke's |
| Blood cultures, CBC | Septic cause |
| EEG | Nonconvulsive status epilepticus; uremic/hepatic changes |
| MRI brain | PRES (posterior T2/FLAIR changes); radiation; structural lesions |
| LP / CSF | Meningitis/encephalitis |
Key Summary
| Type | Key Toxin/Mechanism | Cardinal Sign | Treatment |
|---|---|---|---|
| Hepatic | NH₃, ↑GABA, gut toxins | Asterixis, flap tremor | Lactulose + rifaximin; LT |
| Uremic | Uremic toxins, guanidino compounds | Asterixis, myoclonus | Dialysis / transplant |
| Wernicke's | Thiamine deficiency | Ophthalmoplegia + ataxia + confusion | IV thiamine urgently |
| Septic | Inflammatory mediators, ischemia | Fluctuating consciousness | Treat sepsis |
| Hypertensive / PRES | Cerebral autoregulation failure | Seizures, posterior edema | BP control |
| Radiation | Vascular + demyelinating injury | Cognitive decline, focal deficits | Corticosteroids; bevacizumab |
Parkinsons disease
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive, rich content from Harrison's 22E and Bradley & Daroff. Let me now compile the full response.
Parkinson's Disease
Overview
Parkinson's disease (PD) is the second most common neurodegenerative disorder after Alzheimer's disease, and the most common cause of parkinsonism (~75% of cases). An estimated 10.8 million people worldwide are affected, with the number expected to double within 20 years. Mean age of onset is ~60 years; lifetime risk is ~3% in men and ~2% in women. First described by James Parkinson in 1817, the disease is characterized clinically by motor features resulting from dopaminergic neurodegeneration and pathologically by Lewy body formation. — Harrison's Principles of Internal Medicine 22E
Pathology
Hallmark neuropathological features:
- Degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNc)
- Reduced striatal dopamine
- Lewy bodies and Lewy neurites — intraneuronal proteinaceous inclusions containing α-synuclein (collectively: "Lewy pathology")
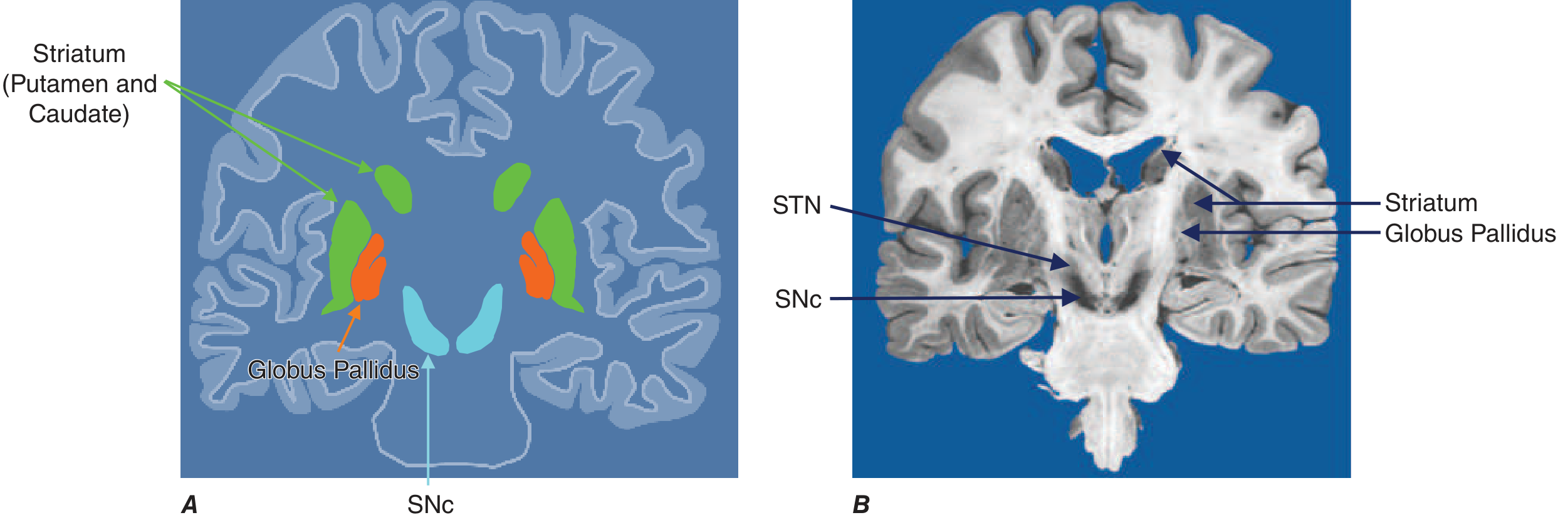
Fig. 446-2 — Basal ganglia nuclei. SNc, substantia nigra pars compacta; STN, subthalamic nucleus. — Harrison's 22E
Neurodegeneration with Lewy pathology is not confined to the dopamine system — it also affects:
- Cholinergic neurons of the nucleus basalis of Meynert (NBM)
- Norepinephrine neurons of the locus coeruleus
- Serotonin neurons in raphe nuclei
- Olfactory system, cerebral hemispheres, spinal cord, and peripheral autonomic nervous system
This explains why PD produces extensive non-motor features beyond dopaminergic deficits.
Braak staging proposes that Lewy pathology begins peripherally — in the GI tract, olfactory system, or dorsal motor nucleus of the vagus — and spreads sequentially to the SNc and then cortex, suggesting the classic motor features emerge at a mid-stage of disease.
Basal Ganglia Circuitry in PD
The classic model: dopamine from SNc inhibits the indirect pathway (via D2 receptors on striatal neurons projecting to GPe) and facilitates the direct pathway (via D1 receptors projecting to GPi).
In PD, dopamine loss causes:
- ↓ inhibition of indirect pathway → ↑ firing of STN and GPi
- ↑ GPi output → excessive inhibition of thalamocortical circuits → reduced cortical activation → bradykinesia, rigidity
This model formed the rationale for DBS targeting the subthalamic nucleus (STN) and globus pallidus internus (GPi).
Clinical Features
| Cardinal Motor Features | Other Motor Features | Nonmotor Features |
|---|---|---|
| Bradykinesia | Micrographia | Anosmia |
| Rest tremor (4–6 Hz) | Masked facies (hypomimia) | Sensory disturbances / pain |
| Rigidity (cogwheel) | Reduced eye blinking | Mood disorders (depression, anxiety, apathy) |
| Postural instability | Drooling, hypophonia | Sleep disturbances (REM sleep behavior disorder) |
| Dysphagia | Autonomic disturbances | |
| Freezing of gait, falls | Orthostatic hypotension | |
| GI disturbances, constipation | ||
| Cognitive impairment / dementia |
Key motor features:
- Bradykinesia: Core feature required for diagnosis; slowing of all voluntary movement; reduced amplitude with repetitive movements
- Rest tremor: Classic "pill-rolling" tremor at 4–6 Hz; suppressed by voluntary movement; typically asymmetric at onset
- Rigidity: Uniform resistance throughout passive range of motion ("lead-pipe"); with superimposed tremor → "cogwheel" rigidity
- Postural instability: Late-stage; contributes heavily to falls
Asymmetry of onset is a key distinguishing feature — onset on one side and gradual spread is strongly supportive of PD vs. other parkinsonian syndromes.
Diagnosis
MDS Clinical Diagnostic Criteria (International Parkinson's and Movement Disorder Society) require motor parkinsonism (bradykinesia + rigidity and/or rest tremor) as the core feature, supported by:
- Supportive criteria (increase confidence): dramatic response to dopaminergic therapy, levodopa-induced dyskinesia, rest tremor, anosmia, cardiac sympathetic denervation on MIBG scintigraphy
- Absolute exclusion criteria: cerebellar signs, downward vertical supranuclear gaze palsy (→ PSP), neuroleptic-induced parkinsonism, lack of expected response to high-dose levodopa
- Red flags requiring counterbalancing supportive criteria: rapid progression to wheelchair within 5 years, early severe autonomic failure, early severe cognitive decline, pyramidal tract signs, bilateral symmetric onset
Two diagnostic levels: clinically established PD and clinically probable PD.
The old UK Brain Bank Criteria (parkinsonism + asymmetry + levodopa response) achieved >90% pathological confirmation. Purely applying tremor + rigidity + bradykinesia had a 24% error rate at postmortem.
Imaging:
- DAT-SPECT / [¹¹C]DTBZ PET: Reduced and asymmetric striatal dopamine transporter uptake, most pronounced in the posterior putamen with relative sparing of caudate — reflecting loss of nigrostriatal terminals
Etiology and Genetics
Most cases are sporadic (cause unknown). ~15% are familial; genetic mutations identified in both dominantly and recessively inherited forms:
| Gene (Designation) | Inheritance | Age of Onset | Notes |
|---|---|---|---|
| SNCA (PARK-SNCA) | AD | Median 46y | α-synuclein — main Lewy body protein; duplications → classical PD |
| LRRK2 (PARK-LRRK2) | AD | Median 56y | Most common genetic form; clinically typical PD |
| GBA1 (PARK-GBA1) | AD (incomplete penetrance) | Variable | Strongest genetic risk factor; faster progression, ↑ dementia risk |
| PRKN / Parkin (PARK-PRKN) | AR | Median 31y | Most common early-onset form; often presents with leg dystonia |
| PINK1 (PARK-PINK1) | AR | Median 32y | Similar to PARK-PRKN; prominent psychiatric features |
| DJ-1 / PARK7 | AR | Median 27y | Clinically very similar to PARK-PRKN |
| VPS35 | AD | Median 52y | Very rare; clinically typical |
Environmental factors: MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine — a mitochondrial toxin in illicit drug manufacturing) produces a PD-like syndrome. Epidemiological associations: pesticides, solvents, rural living, well water — though none proven causal. Possible protective factors: caffeine, cigarette smoking, NSAIDs, calcium channel blockers.
The "double-hit" hypothesis: a combination of a genetic susceptibility variant AND an environmental toxic exposure may be required to cause PD.
Treatment
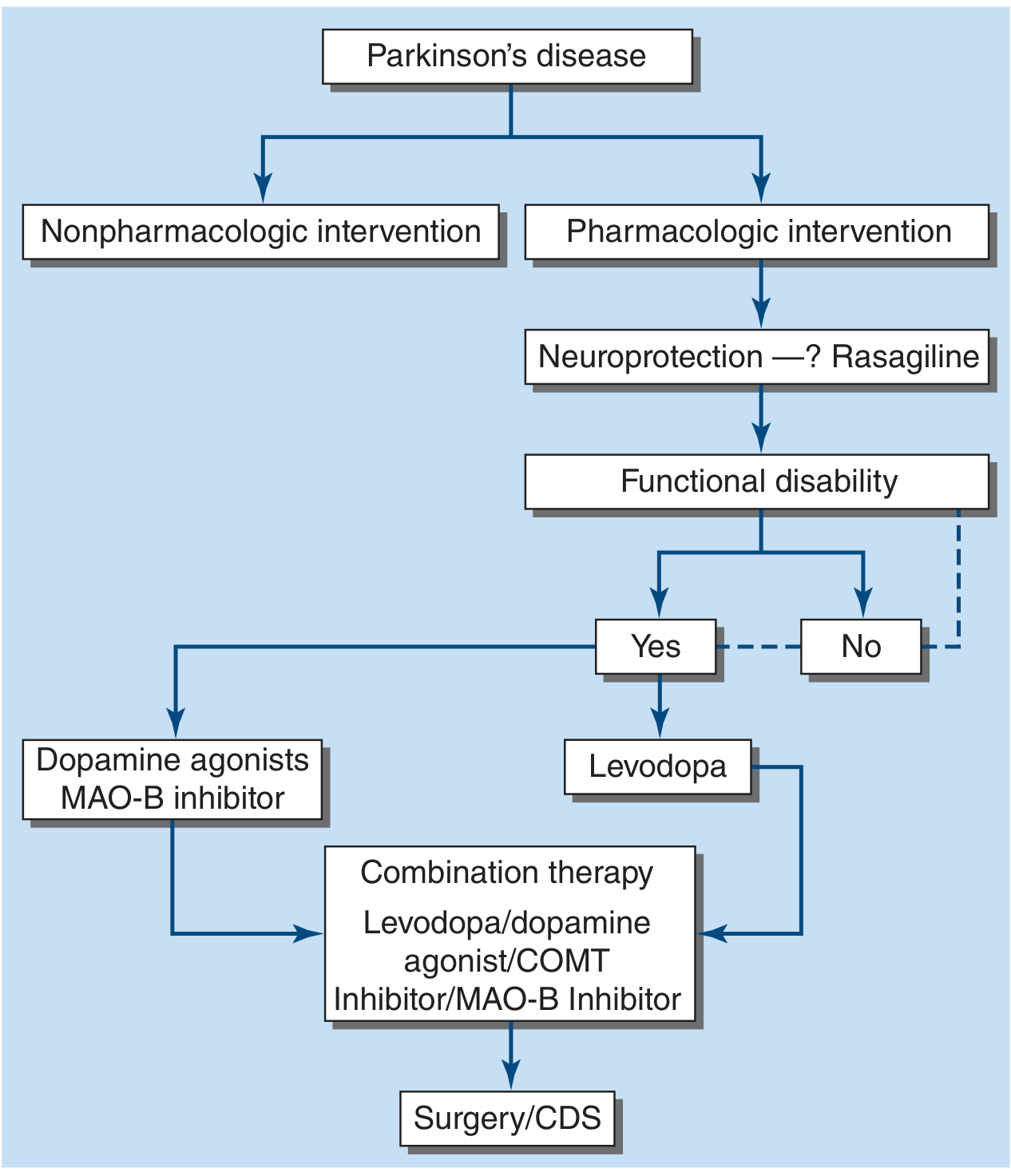
Fig. 446-7 — Treatment options for PD. — Harrison's 22E
1. Levodopa (Gold Standard)
The most effective symptomatic treatment. Levodopa is a dopamine precursor that crosses the BBB. Always combined with a peripheral decarboxylase inhibitor to prevent peripheral conversion to dopamine (which causes nausea, vomiting, orthostatic hypotension):
- Carbidopa/levodopa (Sinemet) — standard in the US
- Benserazide/levodopa (Madopar) — used internationally
- Rytary — extended-release capsule formulation
- Levodopa-carbidopa intestinal gel — continuous intraduodenal infusion for advanced disease
- Inhaled levodopa — absorbed via pulmonary alveoli
Levodopa motor complications (develop in most patients with disease progression):
| Complication | Mechanism | Timing |
|---|---|---|
| Wearing-off | Shorter duration of response as disease progresses | End of dose |
| On-off fluctuations | Unpredictable switches between mobile ("on") and rigid ("off") states | Unpredictable |
| Peak-dose dyskinesias | Involuntary choreiform/dystonic movements at peak plasma level | During "on" phase |
Failure to respond to an adequate levodopa trial should prompt questioning of the PD diagnosis.
2. Dopamine Agonists
Act directly on striatal dopamine receptors; may be used as monotherapy in early disease or adjuncts to levodopa. Reduce "off" time when used with levodopa. Greater tendency to cause impulse control disorders (gambling, hypersexuality), hallucinations, and somnolence than levodopa, especially in elderly patients.
3. MAO-B Inhibitors
Selegiline, rasagiline (irreversible) and safinamide (reversible, also blocks sodium channels and inhibits glutamate). Block central dopamine metabolism → ↑ synaptic dopamine. Used as:
- Monotherapy in early PD
- Adjunct to levodopa to reduce "off" time
Risk: may increase dyskinesia (manage by reducing levodopa dose). No cheese effect at therapeutic doses (selective for MAO-B; does not inhibit gut MAO-A). Theoretical serotonin syndrome risk with SSRIs — rare in practice.
4. COMT Inhibitors
Entacapone (with each levodopa dose), tolcapone (3×/day), opicapone (once daily — most recently approved). Block peripheral COMT metabolism of levodopa → ↑ elimination half-life → ↑ brain levodopa availability; reduce "off" time. Combination tablet: Stalevo (levodopa + carbidopa + entacapone). Key side effects: dopaminergic (nausea, dyskinesia); rare fatal hepatic toxicity with tolcapone (monitor LFTs); severe diarrhea in 5–10%.
5. Deep Brain Stimulation (DBS)
Indicated in patients with motor fluctuations and dyskinesias not adequately controlled by medical therapy. Targets:
- STN (subthalamic nucleus): Larger benefit in "off" state; allows greater levodopa dose reduction; slightly higher neuropsychiatric risk
- GPi (globus pallidus internus): Better dyskinesia suppression; better long-term flexibility; relatively safer neuropsychiatric risk profile; preferred for "brittle" dyskinesia
Both STN and GPi DBS have shown similar overall motor outcomes across multiple RCTs. The German Parkinson Study Group 2006 RCT demonstrated improved quality of life with bilateral STN-DBS vs. best medical therapy in advanced PD patients <75 years.
6. Nonpharmacologic Interventions
- Physiotherapy and exercise (neuroprotective potential being studied)
- Speech therapy (hypophonia, dysphagia)
- Occupational therapy
- Dietetic support (aspiration risk, weight loss)
Nonmotor Features & Management
| Feature | Management |
|---|---|
| Depression / anxiety | SSRIs, SNRIs; pramipexole has some antidepressant effect |
| Dementia (PDD) | Rivastigmine (only FDA-approved); donepezil used off-label |
| Psychosis / hallucinations | Reduce dopaminergic medications; quetiapine or clozapine (avoid typical antipsychotics) |
| REM sleep behavior disorder (RBD) | Clonazepam or melatonin; often precedes motor PD by years |
| Orthostatic hypotension | Fludrocortisone, midodrine, droxidopa |
| Constipation | Fiber, hydration, laxatives; probiotics being studied |
| Dysphagia / sialorrhea | Speech therapy, modified diet; botulinum toxin for drooling |
Differential Diagnosis of Parkinsonism
| Condition | Key Distinguishing Features |
|---|---|
| Essential tremor | Action/postural tremor (not rest); responds to alcohol; head tremor |
| Multiple system atrophy (MSA) | Early autonomic failure, cerebellar ataxia, or pyramidal signs; poor levodopa response |
| Progressive supranuclear palsy (PSP) | Vertical gaze palsy (downward), early falls, axial rigidity, frontal dementia |
| Corticobasal degeneration (CBD) | Alien limb, apraxia, cortical sensory loss, asymmetric |
| Lewy body dementia (DLB) | Cognitive decline before or within 1 year of parkinsonism; fluctuating cognition, visual hallucinations |
| Drug-induced parkinsonism | History of dopamine antagonists (antipsychotics, metoclopramide); symmetric onset |
| Vascular parkinsonism | Lower-body predominance, gait disorder, history of cerebrovascular disease |
Prognosis
PD is progressive but not uniformly fatal. Levodopa therapy has dramatically improved survival and quality of life. Disease progression leads to:
- Motor complications from levodopa (most patients within 5–10 years)
- Nondopaminergic features (falls, freezing, dementia, autonomic failure) which become the primary source of late-stage disability and main drivers of nursing home placement
- Long-term cognitive decline / PD dementia in ~80% by 20 years
SARS-CoV-2 infection worsens PD features and increases complication risk; new-onset PD cases following COVID-19 have been reported, paralleling cases associated with the 1918 influenza epidemic. — Harrison's Principles of Internal Medicine 22E; Bradley and Daroff's Neurology in Clinical Practice
This is a shared conversation. Sign in to Orris to start your own chat.