Porphyria
Reading File
Finding Sources
Searching PubMed
porphyria
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Loading Image
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
I now have everything needed for a thorough, well-illustrated answer. Here is the full response:
Porphyria
The porphyrias are a group of rare inherited (occasionally acquired) metabolic disorders caused by defects in the heme biosynthesis pathway. Each type reflects a specific enzyme deficiency, leading to the accumulation of toxic porphyrin intermediates upstream of the block. The name comes from the Greek for "purple," referring to the red-purple pigmentation these intermediates impart to urine. - Lippincott's Illustrated Reviews: Biochemistry, 8e
Heme Synthesis Pathway and Enzyme Defects
The diagram below maps the entire heme pathway, showing which enzyme is deficient in each type of porphyria and whether the resulting accumulation is hepatic or erythropoietic:
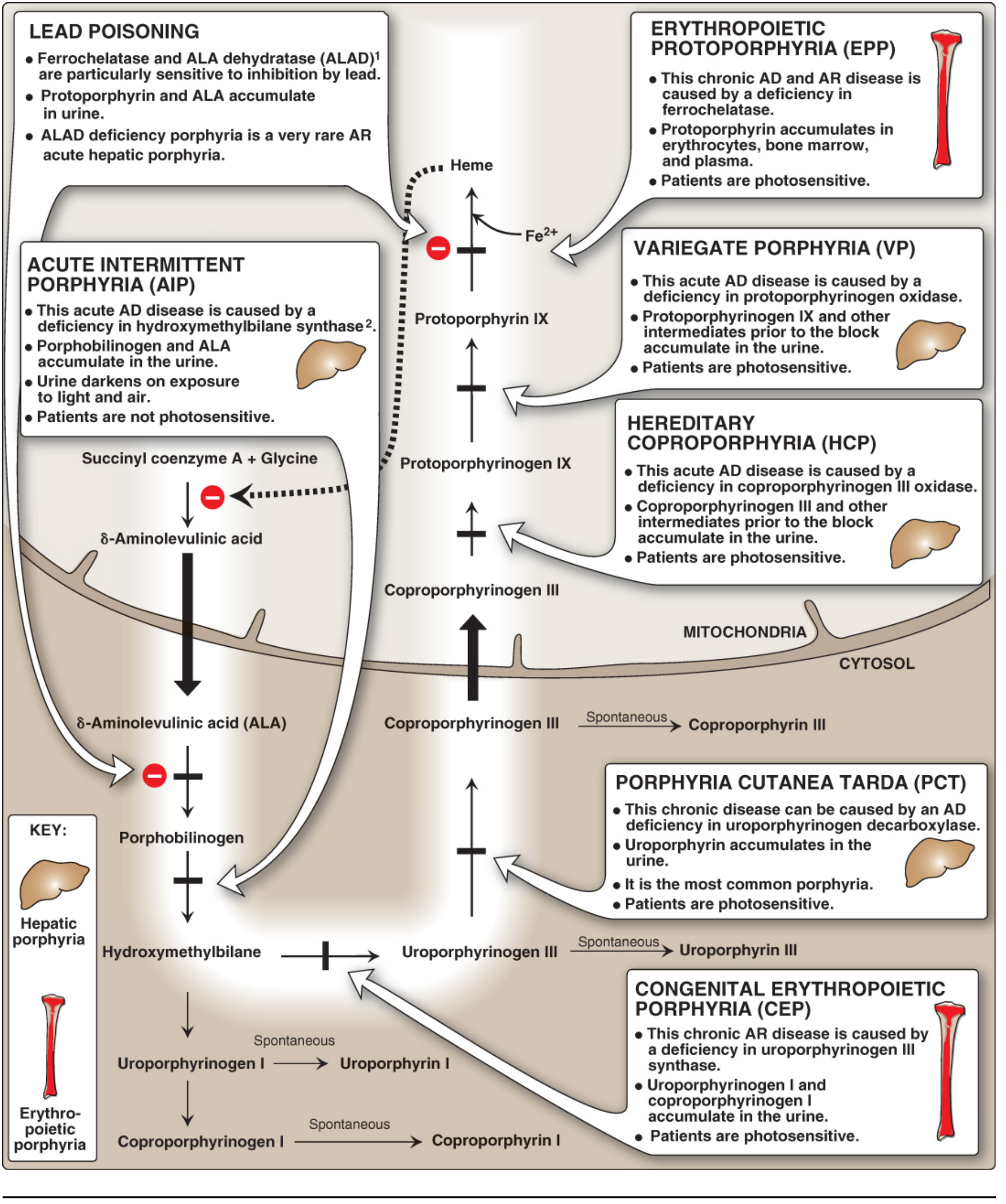
The key principle: defects early in the pathway (before tetrapyrrole ring formation) cause ALA and PBG accumulation, producing abdominal and neuropsychiatric symptoms. Defects later in the pathway (after ring formation) cause accumulation of photosensitizing tetrapyrrole intermediates, producing cutaneous photosensitivity. - Lippincott's Biochemistry, 8e
Classification
Porphyrias are classified by:
- Site of enzyme defect: hepatic vs. erythropoietic
- Clinical behavior: acute (neurovisceral attacks) vs. non-acute (cutaneous only)
| Type | Enzyme Deficient | Inheritance | Major Features |
|---|---|---|---|
| ALA dehydratase deficiency porphyria (ADP) | ALA dehydratase (ALAD) | AR | Acute; very rare |
| Acute Intermittent Porphyria (AIP) | Hydroxymethylbilane synthase (HMBS / PBG deaminase) | AD | Acute neurovisceral; no skin |
| Porphyria Cutanea Tarda (PCT) | Uroporphyrinogen decarboxylase (UROD) | AD (or acquired) | Most common porphyria; skin only |
| Hereditary Coproporphyria (HCP) | Coproporphyrinogen oxidase (CPO) | AD | Acute + skin (mild) |
| Variegate Porphyria (VP) | Protoporphyrinogen oxidase (PPOX) | AD | Acute + skin (PCT-like) |
| Congenital Erythropoietic Porphyria (CEP) | Uroporphyrinogen III synthase | AR | Severe photosensitivity from infancy |
| Erythropoietic Protoporphyria (EPP) | Ferrochelatase | AD/AR | Photosensitivity; risk of liver disease |
| X-linked Protoporphyria (XLEPP) | ALAS2 gain-of-function | X-linked | EPP-like |
Acute Hepatic Porphyrias (AIP, HCP, VP, ADP)
Pathogenesis
In the hepatic porphyrias, reduced heme synthesis removes the normal negative feedback on ALAS1. This derepresses ALAS1, driving overproduction of ALA and PBG. ALA accumulation is neurotoxic and is responsible for most of the neurologic sequelae. - Lippincott's Biochemistry, 8e
Precipitating Factors
- Drugs metabolized by cytochrome P450 (e.g., barbiturates, sulfonamides, rifampicin, oral contraceptives containing progestins)
- Hormonal shifts - menstrual cycle, pregnancy
- Dietary restriction / fasting (reduces heme synthesis)
- Infections, alcohol, stress
Clinical Presentation
Attacks typically begin with severe colicky abdominal pain, followed by:
- Neuropsychiatric: agitation, hallucinations, anxiety, delirium, seizures
- Motor neuropathy: ascending weakness mimicking Guillain-Barré syndrome (GBS); can be proximal or distal, asymmetric, may involve face/bulbar musculature
- Autonomic: tachycardia, hypertension, pupillary dilation, constipation, urinary retention, incontinence
- Hyponatremia: due to SIADH
- Urine: brown or port-wine colored; darkens on exposure to light and air (ALA/PBG oxidation)
On nerve conduction studies: markedly reduced CMAP amplitudes with signs of axonal degeneration on needle EMG. CSF protein is normal or mildly elevated. - Harrison's 22e
AIP has no photosensitivity. HCP and VP add photosensitive blistering skin lesions to the neurovisceral picture.
Laboratory Diagnosis
- Urine ALA and PBG: elevated during attacks (the screening test of choice)
- Specific enzyme activity in erythrocytes or leukocytes
- Stool porphyrins: elevated fecal coproporphyrins in HCP and VP
- VP has a characteristic plasma fluorescence at 626 nm; HCP at 619 nm
Treatment of Acute Attacks
- Remove precipitants - stop offending drugs, treat infections
- IV glucose 10-20 g/h - reduces ALAS1 induction; adequate for mild attacks
- IV hemin (hematin) 2-5 mg/kg/day for 3-14 days - replenishes heme, suppresses ALAS1; used if no improvement within 24 h
- Givosiran (Givlaari) - a subcutaneous siRNA that silences hepatic ALAS1 mRNA; 2.5 mg/kg monthly; shown in clinical trials to reduce attack frequency, lower urinary ALA/PBG, improve pain scores and quality of life; approved for recurrent acute intermittent porphyria - Harrison's 22e
- Supportive: opioids for pain, beta-blockers for autonomic instability, benzodiazepines for seizures
Porphyria Cutanea Tarda (PCT) - Most Common
PCT is the most common porphyria, caused by deficiency of uroporphyrinogen decarboxylase (UROD) in the liver. Only ~20% have germline UROD mutations (Type II); the remainder have acquired enzymatic inhibition by iron-generated oxidants in the liver (Type I). - Lippincott's Biochemistry, 8e
Risk/triggering factors: hepatic iron overload, excess alcohol, estrogen therapy, hepatitis B or C, HIV infection, hemochromatosis (HFE mutations), halogenated hydrocarbons.
Clinical features (onset typically 4th-5th decade):
- Blisters, erosions, and crusts on sun-exposed areas (dorsal hands most common)
- Skin fragility
- Hypertrichosis (face, temporal area)
- Hyperpigmentation and milia
- Scleroderma-like changes
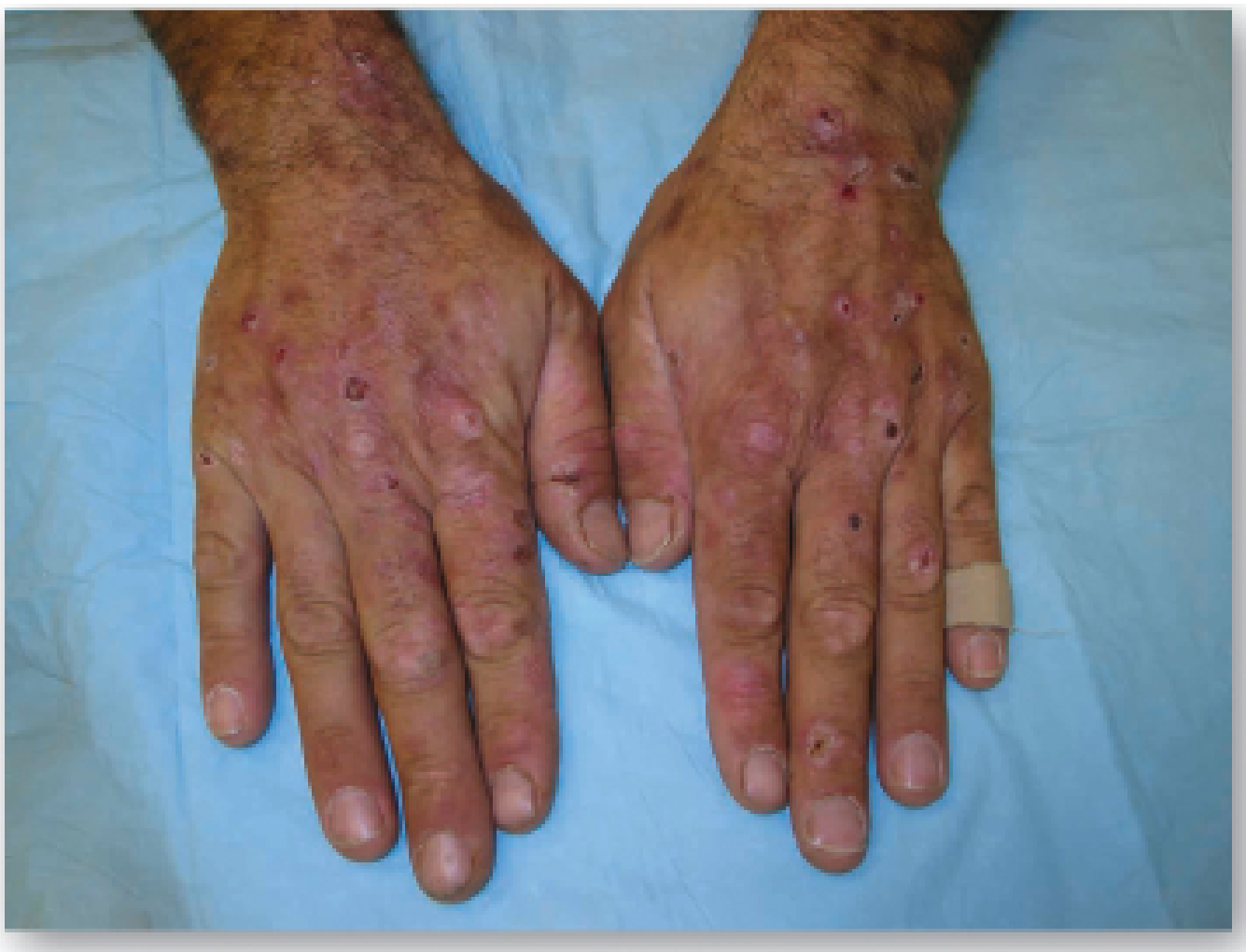
Biochemistry: urine uroporphyrin markedly elevated (>uroporphyrin I predominates); normal urine PBG distinguishes PCT from acute porphyrias.
Treatment:
- Eliminate triggers (stop alcohol, estrogen, iron supplements)
- Phlebotomy (450 mL every 2 weeks) to reduce hepatic iron overload - allows new UROD to be synthesized
- Low-dose hydroxychloroquine (100-200 mg twice weekly) - equally effective to phlebotomy; chelates and mobilizes uroporphyrins from liver; used when phlebotomy is contraindicated (e.g., anemia)
- Treat underlying hepatitis C - Yamada's Gastroenterology, 7e; Fitzpatrick's Dermatology
Variegate Porphyria (VP)
VP combines features of both PCT and AIP - it is also known as "South African genetic porphyria" because of a founder mutation (R59W in PPOX) highly prevalent in Afrikaner populations. The deficient enzyme is protoporphyrinogen oxidase (PPOX). - Andrews' Diseases of the Skin
- 40-70% have skin symptoms (PCT-like blistering)
- 27% have acute neurovisceral attacks
- 14% have both simultaneously
- Key diagnostic finding: plasma fluorescence at 626 nm (unique to VP)
- Fecal coproporphyrins and protoporphyrins are always elevated
- Urinary coproporphyrin > uroporphyrin (distinguishes from PCT)
VP patients (like HCP and AIP patients) have an increased long-term risk of hepatocellular carcinoma - liver imaging is recommended after age 50. - Andrews' Diseases of the Skin
Erythropoietic Porphyrias
Congenital Erythropoietic Porphyria (CEP - Günther's disease)
- AR deficiency of uroporphyrinogen III synthase
- Most severe porphyria; presents in infancy with dark urine and photosensitivity
- Uroporphyrinogen I and coproporphyrinogen I accumulate (isomer I has no physiological role)
- Features: vesicles/bullae on sun-exposed skin, sclerodermoid scarring, hypertrichosis, red fluorescent teeth (erythrodontia), hemolytic anemia
- Treatment: strict sun protection, blood transfusions; bone marrow transplantation may be curative
Erythropoietic Protoporphyria (EPP)
- Deficiency of ferrochelatase (AD with reduced penetrance, or AR)
- Protoporphyrin accumulates in erythrocytes, bone marrow, and plasma
- Presents in early childhood with acute painful photosensitivity (burning, itching within minutes of sun exposure) - no blistering
- Risk of cholestatic liver disease from protoporphyrin accumulation
- β-carotene (provitamin A) and afamelanotide (an alpha-MSH analog) are used for photoprotection
Hepatoerythropoietic Porphyria (HEP)
HEP is the homozygous form of PCT (homozygous or compound heterozygous UROD deficiency, with activity ~10% of normal). Clinical features resemble CEP but are distinguished biochemically by elevated erythrocyte protoporphyrins + urinary uroporphyrins. - Andrews' Diseases of the Skin
Pseudoporphyria
Not a true porphyria. Patients develop PCT-like blistering with normal porphyrin levels. Caused by NSAIDs (especially naproxen), voriconazole, tetracyclines, imatinib, tanning beds, and hemodialysis. Treatment: discontinue the offending agent and sun protection. - Andrews' Diseases of the Skin
Key Distinguishing Points (Summary)
| Feature | AIP | PCT | VP | HCP | EPP |
|---|---|---|---|---|---|
| Neurovisceral attacks | Yes | No | Yes | Yes | No |
| Photosensitivity/skin | No | Yes | Yes | Mild | Yes (acute burning) |
| Urine PBG elevated | During attacks | Normal | During attacks | During attacks | Normal |
| Key diagnostic test | Urine ALA/PBG | Urine uroporphyrin | Plasma fluor. 626 nm | Fecal coproporphyrin | Erythrocyte protoporphyrin |
| Inheritance | AD | AD/acquired | AD | AD | AD/AR |
Recent Evidence (PubMed 2024-2026)
A 2024 meta-analysis on acute hepatic porphyria and hepatocellular carcinoma (PMID 39438413) confirmed that patients with acute hepatic porphyria have a significantly elevated risk of HCC, supporting current recommendations for regular liver surveillance after age 50 in all acute porphyria patients.
Sources: Harrison's Principles of Internal Medicine 22e; Lippincott's Illustrated Reviews: Biochemistry 8e; Andrews' Diseases of the Skin; Fitzpatrick's Dermatology; Yamada's Textbook of Gastroenterology 7e; Bradley and Daroff's Neurology in Clinical Practice
This is a shared conversation. Sign in to Orris to start your own chat.