Please teach me about cystic fibrosis
Reading File
Finding Sources
Searching PubMed
"cystic fibrosis"[MeSH Terms] AND "CFTR modulators"
Reading File
Reading File
Searching Images
cystic fibrosis CFTR protein chloride channel pathophysiology
Reading File
Reading File
Loading Image
Reading File
Loading Image
Finding Sources
Reading File
Reading File
I now have comprehensive, authoritative information to write a thorough educational overview of cystic fibrosis. Here it is:
Cystic Fibrosis — A Comprehensive Overview
1. What Is Cystic Fibrosis?
Cystic fibrosis (CF) is the most common lethal autosomal recessive genetic disorder in populations of European ancestry, occurring in approximately 1 in 2,500 births (carrier frequency ~1 in 25). It is caused by mutations in the CFTR gene (Cystic Fibrosis Transmembrane Conductance Regulator), which encodes a regulated chloride channel in the apical membrane of epithelial cells. The disease primarily affects the lungs, exocrine pancreas, intestine, hepatobiliary system, and male reproductive tract.
It is far less common in other populations: ~1 in 15,000 in Black individuals and ~1 in 31,000 in Asian individuals.
2. The CFTR Protein
The CFTR gene spans ~190 kb with 27 exons, encoding a large integral membrane protein of ~170 kDa. It belongs to the ABC (ATP-binding cassette) family of transport proteins and has five domains:
| Domain | Function |
|---|---|
| Two membrane-spanning domains (12 TM segments) | Form the chloride channel pore |
| Two nucleotide-binding domains (NBD1, NBD2) | Bind and hydrolyze ATP to open/close the channel |
| Regulatory (R) domain | Contains phosphorylation sites; regulates channel activity |
Thompson & Thompson Genetics and Genomics in Medicine, 9th ed.
3. Pathophysiology
The loss of CFTR function disrupts fluid and electrolyte transport across epithelial surfaces throughout the body:
- Sweat glands: CFTR normally reabsorbs chloride in the duct. Without it, chloride (and consequently sodium) cannot be reabsorbed → elevated sweat sodium and chloride (the diagnostic hallmark).
- Lungs: Hyperabsorption of sodium + reduced chloride secretion → depletion of airway surface liquid → thick, adherent mucus → disrupted mucociliary clearance → chronic infection (especially Pseudomonas aeruginosa).
- Pancreas: Inspissated secretions block pancreatic ductules → loss of lipase, trypsin, and chymotrypsin secretion → maldigestion and steatorrhea.
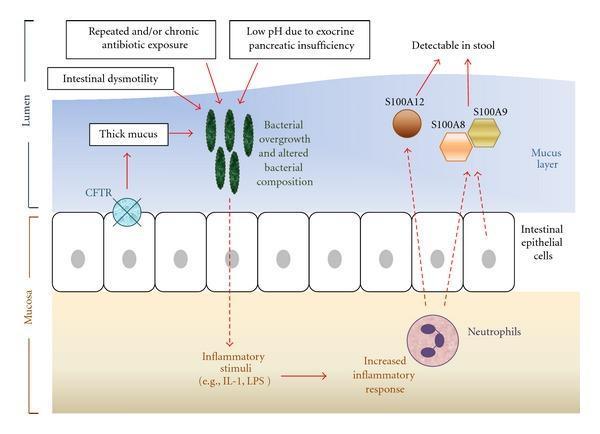
- Intestine: Thick mucus, dysmotility, altered microbiome, and low pH from pancreatic insufficiency promote bacterial overgrowth and intestinal inflammation.
4. Classes of CFTR Mutations
Over 2,000 CFTR sequence variants have been identified. They are grouped into 6 functional classes, each requiring a different therapeutic approach:
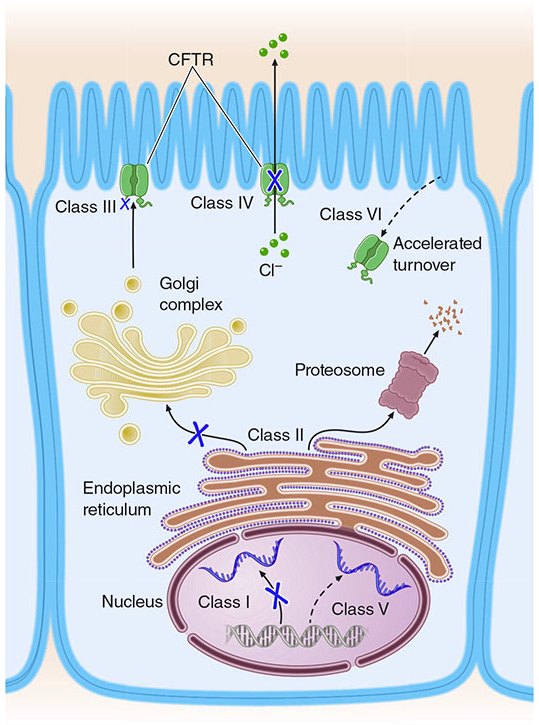
| Class | Defect | Example | Mechanism |
|---|---|---|---|
| I | No protein made | W1282X | Premature stop codon → unstable mRNA |
| II | Misfolded protein, ER retention | F508del (~70% of all CF alleles) | Protein degraded by proteasome before reaching cell surface |
| III | Protein reaches surface but channel won't open | G551D | Defective ATP-binding/gating |
| IV | Reduced chloride conductance | R117H | Channel opens but conducts Cl⁻ poorly |
| V | Reduced amount of normal CFTR | Splice site variants | Reduced mRNA production |
| VI | Normal protein but accelerated turnover | Q1412X | Rapid degradation at cell surface |
Thompson & Thompson Genetics and Genomics in Medicine, 9th ed., p. 284–285
The F508del mutation (deletion of phenylalanine at position 508 in NBD1) is a Class II variant — it is the most clinically important, accounting for ~70% of all CF alleles in European populations. ~50% of patients are homozygous F508del.
5. Clinical Manifestations
CF is a multi-organ disease. The diagram below shows the intestinal inflammation mechanism:
Pulmonary (most common cause of morbidity and mortality)
- Chronic productive cough, bronchitis, bronchiectasis
- Recurrent pneumonia (Staphylococcus aureus early; Pseudomonas aeruginosa chronically)
- Air trapping, emphysema, atelectasis
- Sinusitis, nasal polyps
- Eventual respiratory failure and cor pulmonale (right heart failure)
Pancreatic
- 85–90% of patients have exocrine pancreatic insufficiency (maldigestion, steatorrhea, fat-soluble vitamin deficiency — A, D, E, K)
- CF-related diabetes mellitus (CFRD): 4–7% of all patients; more common in adults
- Pancreatitis (1–2%)
- ~10–15% have pancreatic sufficiency (better prognosis; associated with milder CFTR mutations)
Gastrointestinal
- Meconium ileus: intestinal obstruction in neonates, 10–25% of CF newborns
- Distal intestinal obstruction syndrome (DIOS): fecal impaction in adults (~18%)
- Rectal prolapse, intussusception
- GERD (very common in adults, ~80%)
Hepatobiliary
- Focal biliary cirrhosis (2–3% overall; up to 11–70% in adults)
- Portal hypertension, esophageal varices
- Gallstones (8–25%), atrophic gallbladder
Reproductive
- Males: >98% are infertile due to congenital bilateral absence of the vas deferens (CBAVD) — sperm production is normal; assisted fertilization via testicular sperm aspiration is possible
- Females: reduced fertility due to increased mucus viscosity, but most can achieve pregnancy
Sleisenger and Fordtran's Gastrointestinal and Liver Disease; The Harriet Lane Handbook, 23rd ed.
6. Diagnosis
| Test | Interpretation |
|---|---|
| Sweat chloride (pilocarpine iontophoresis) | ≥60 mmol/L = positive; gold standard |
| Newborn screening (IRT/DNA) | >60% of US diagnoses now made via newborn screen |
| CFTR genetic analysis (127-variant panel + F508del) | Confirms diagnosis; identifies compound heterozygotes |
| Nasal potential difference (NPD) | Used when sweat test equivocal |
The median age at diagnosis in the US is 4 months, with 67% diagnosed in the first year of life. About 10% are diagnosed after age 10.
7. Treatment
Airway Clearance
- Chest physiotherapy (percussion, vibration)
- Hypertonic saline (inhaled) — restores airway surface liquid
- Dornase alfa (Pulmozyme) — DNase that cleaves DNA in thick mucus, reducing viscosity
Antimicrobials
- Inhaled tobramycin or aztreonam for P. aeruginosa suppression
- Oral azithromycin (anti-inflammatory + antimicrobial properties)
CFTR Modulator Therapy — A Revolution in CF Care
These drugs target the underlying molecular defect and have dramatically improved outcomes:
| Brand Name | Drug(s) | Mechanism | Eligible Patients |
|---|---|---|---|
| Kalydeco | Ivacaftor | Potentiator — opens defective channels at cell surface | G551D and 96 other gating/surface mutations; age ≥4 months |
| Orkambi | Lumacaftor + ivacaftor | Corrector + Potentiator | Homozygous F508del; age ≥2 years |
| Symdeko | Tezacaftor + ivacaftor | Corrector + Potentiator | F508del homozygous or one of 154 mutations; age ≥6 years |
| Trikafta | Elexacaftor + tezacaftor + ivacaftor | Dual corrector + Potentiator | ≥1 copy of F508del or 177 mutations; age ≥6 years |
Trikafta (ETI triple therapy) is now the standard of care for eligible patients (~90% of CF patients qualify). It significantly improves FEV₁, reduces exacerbations, and improves quality of life. A recent network meta-analysis (2025) confirmed ETI as the most efficacious CFTR modulator combination.
Murray & Nadel's Textbook of Respiratory Medicine; The Harriet Lane Handbook, 23rd ed.
Nutritional Support
- Pancreatic enzyme replacement therapy (PERT) before meals
- Fat-soluble vitamins (A, D, E, K) supplementation
- High-calorie diet; maintain BMI ≥50th percentile in children
Advanced / End-Stage
- Lung transplantation for end-stage lung disease
- Gene therapy remains investigational — mRNA-based and viral vector strategies are under active development
8. Prognosis
Outcomes have dramatically improved over the past decades. Prior to CFTR modulators, median survival was ~30 years. With current therapy including Trikafta, median predicted survival now exceeds 48 years, and patients diagnosed today in high-income countries with access to modulator therapy may have near-normal life expectancy. Pulmonary disease remains the leading cause of death.
9. Genetics — Screening & Counseling Key Points
- Carrier frequency: ~1 in 25 in people of European ancestry
- Inheritance: autosomal recessive — both parents must carry a mutant allele; 25% chance of affected child per pregnancy
- Genotype–phenotype correlations: CFTR genotype predicts pancreatic function well, but is a poor predictor of lung disease severity (modulated by genes like TGFβ1, IL-8, IFRD1)
- Population screening: universal carrier screening offered in the US, Canada, Australia, most of Western Europe
- Prenatal diagnosis: DNA analysis at 10–12 weeks via chorionic villus sampling; preimplantation genetic testing (PGT) available after IVF
Thompson & Thompson Genetics and Genomics in Medicine, 9th ed., p. 285–286
Summary Table
| Feature | Detail |
|---|---|
| Inheritance | Autosomal recessive |
| Gene | CFTR (chromosome 7q31) |
| Most common mutation | F508del (~70% of European alleles) |
| Primary defect | Defective Cl⁻ channel → thick secretions |
| Hallmark diagnostic test | Sweat chloride ≥60 mmol/L |
| Most common cause of death | Respiratory failure from chronic infection/bronchiectasis |
| Revolutionary treatment | Trikafta (elexacaftor/tezacaftor/ivacaftor) |
| Current median survival | >48 years (with modern therapy) |
Recent evidence note: A 2025 network meta-analysis (PMID 40102290) confirms ETI (elexacaftor/tezacaftor/ivacaftor) is the most efficacious CFTR modulator. A 2025 systematic review (PMID 40399734) examines pharmacokinetics of modulators in special populations (pregnancy, transplant, etc.), highlighting that dosing guidance is still evolving.
This is a shared conversation. Sign in to Orris to start your own chat.