
IMG_0128.jpg
Explain this
Reading File
Finding Sources
analyse_media
Here is a full breakdown of everything in this image, which covers the Collecting Tubule section from First Aid for the USMLE Step 1.
The Collecting Tubule - Complete Explanation
Overview
The collecting tubule is the final fine-tuning segment of the nephron. It handles the last 3-5% of filtered Na+, but its hormonal regulation makes it the most clinically important segment for managing blood pressure, potassium balance, and acid-base status.
1. Aldosterone - The Master Regulator
Aldosterone is a steroid mineralocorticoid hormone produced by the adrenal cortex (zona glomerulosa).
Mechanism:
- Being lipid-soluble, it crosses the cell membrane and binds to the mineralocorticoid receptor (MR) inside the cell
- The receptor-ligand complex enters the nucleus and drives mRNA transcription → new protein synthesis
- These newly synthesized proteins are what actually do the work (this is why aldosterone's effects take 30-60 minutes - it needs to make new proteins)
2. Effects in Principal Cells (Na+/K+ Regulation)
Principal cells are the main workhorses of the collecting tubule. Aldosterone upregulates three things here:
| Target | Effect | Result |
|---|---|---|
| ENaC (epithelial Na+ channel) | More channels on the apical/luminal membrane | More Na+ enters from lumen into cell |
| Na+/K+ ATPase | More pumps on basolateral membrane | Na+ pumped into blood, K+ pumped into cell |
| Apical K+ conductance (ROMK channels) | More K+ channels on apical membrane | K+ secreted into urine |
The key cascade:
- ENaC reabsorbs Na+ (positive ions) from the lumen
- This leaves the lumen negatively charged (lumen negativity)
- That negative charge electrostatically pulls K+ out into the lumen - this is how aldosterone causes K+ wasting
- Net result: Na+ retained in blood, K+ lost in urine
This is why hyperaldosteronism causes hypertension (Na+ retention) + hypokalemia (K+ wasting), and why aldosterone blockers (spironolactone, eplerenone) are potassium-sparing diuretics.
3. Effects in Alpha-Intercalated Cells (Acid-Base Regulation)
Alpha-intercalated cells handle H+ secretion. They respond to the lumen negativity created by principal cells:
- Lumen negativity → stimulates apical H+ ATPase to pump H+ into the urine
- Inside the cell, carbonic anhydrase produces H+ + HCO3-
- H+ goes into the lumen (acidifies urine)
- HCO3- exits via the basolateral HCO3-/Cl- exchanger → enters the blood
Net effect: Urine is acidified, blood HCO3- rises (metabolic alkalosis tendency)
This explains why hyperaldosteronism causes metabolic alkalosis - both excess H+ secretion here and hypokalemia drive HCO3- retention.
4. ADH (Antidiuretic Hormone / Vasopressin)
ADH acts independently of aldosterone to control water reabsorption:
- Binds to V2 receptors on the basolateral side of principal cells
- Triggers cAMP → protein kinase A signaling cascade
- This causes aquaporin-2 (AQP2) water channels to be inserted into the apical (lumen-facing) membrane
- Water then flows osmotically from dilute tubular fluid into the hypertonic medullary interstitium → concentrated urine
ADH controls how much water is reabsorbed. Aldosterone controls how much Na+ is reabsorbed. These two hormones together determine final urine volume and concentration.
5. The Last Details
- 3-5% Na+ reabsorbed here - small fraction but tightly regulated (this is where all the fine-tuning happens)
- Urea reabsorption occurs only in the medullary collecting duct (NOT the cortical part). ADH stimulates urea transporters (UT-A1/UT-A3) in the medullary duct, which recycles urea back into the medullary interstitium - this is essential for building the concentration gradient needed to concentrate urine.
Clinical Summary Table
| Hormone | Receptor | Cell Type | Key Action | Clinical Implication |
|---|---|---|---|---|
| Aldosterone | Mineralocorticoid (intracellular) | Principal cells | ↑ENaC, ↑Na+/K+ ATPase, ↑K+ secretion | Hyperaldo = HTN + hypokalemia + met. alkalosis |
| Aldosterone | Same | Alpha-intercalated cells | ↑H+ ATPase → H+ secretion | Excess → metabolic alkalosis |
| ADH | V2 (basolateral) | Principal cells | Insert AQP2 on apical side | SIADH = hyponatremia; Diabetes insipidus = dilute urine |
Can you show a propee image shiwing cells blood lumen
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Here are two proper textbook diagrams showing the Principal Cell of the collecting tubule with Lumen on the left and Interstitium (blood side) on the right:
Diagram 1 - Principal Cell (Medical Physiology)
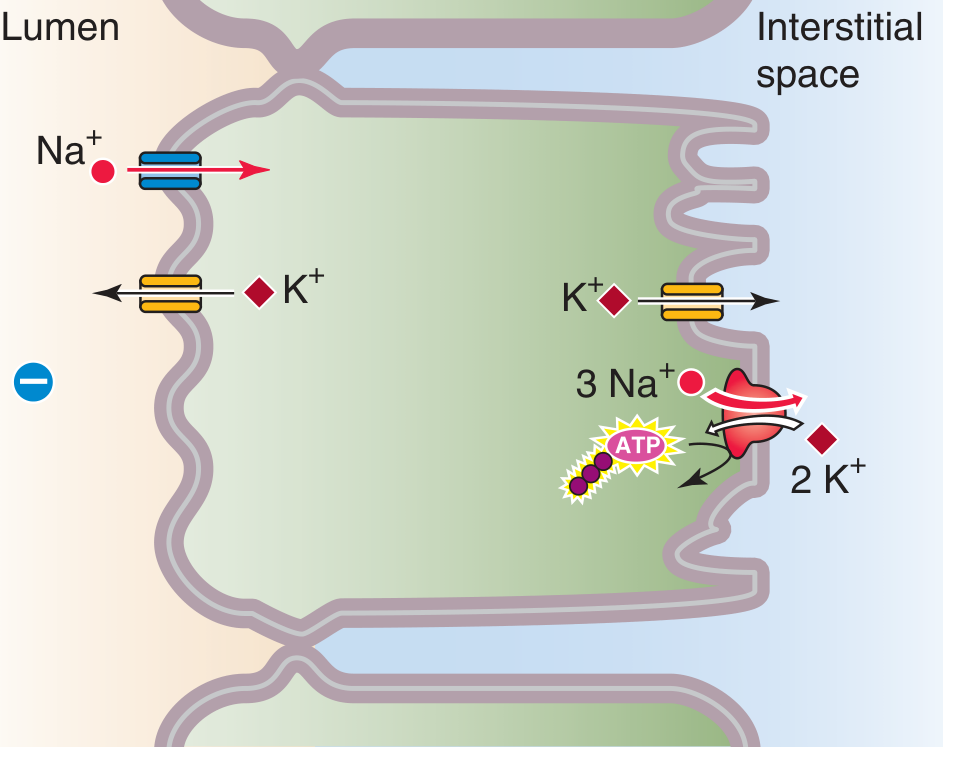
Diagram 2 - Na+ Transport in the Cortical Collecting Duct Principal Cell (Comprehensive Clinical Nephrology)
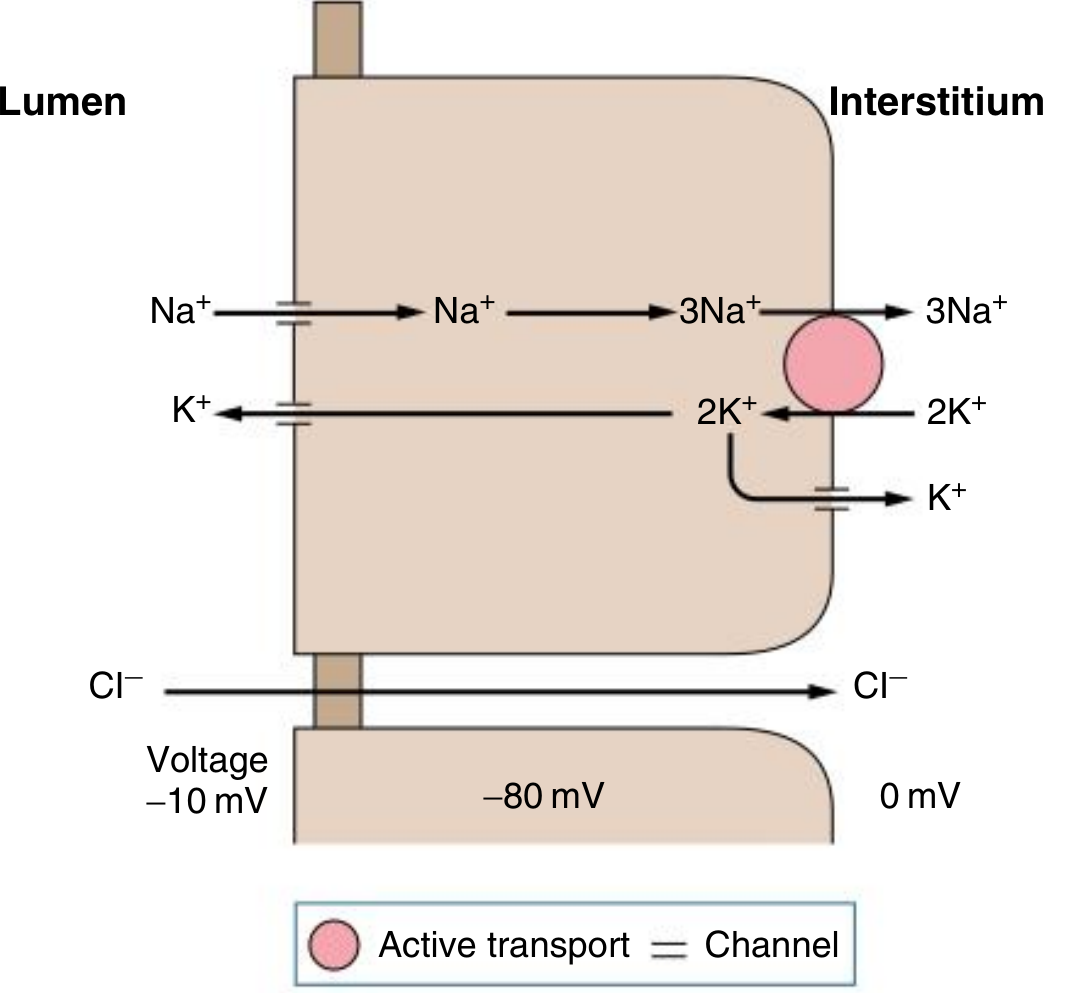
How to Read These Diagrams
Left side = Lumen (inside the tubule, where filtered urine flows)
Middle = Principal Cell (the tubular epithelial cell)
Right side = Interstitium/Blood (peritubular capillaries)
What's happening step by step:
-
Apical (lumen-facing) membrane:
- Na+ enters the cell passively through ENaC channels (down its electrochemical gradient - lumen → cell)
- K+ exits the cell into the lumen through apical K+ channels (ROMK) - this is K+ secretion/wasting
-
Basolateral (blood-facing) membrane:
- The Na+/K+ ATPase pump (pink circle = active transport using ATP) pumps 3 Na+ out into the interstitium and pulls 2 K+ in from the blood
- This keeps intracellular Na+ low (driving apical ENaC entry) and intracellular K+ high (driving apical K+ secretion)
- Some K+ also exits basally via basolateral K+ channels
-
Voltage:
- Diagram 1 shows the voltages: Lumen = -10 mV, Cell interior = -80 mV, Interstitium = 0 mV
- The lumen is negatively charged relative to the interstitium - this is "lumen negativity" driven by the net movement of positive Na+ ions out of the lumen
- This lumen negativity is what electrostatically drives K+ and H+ secretion into the urine
-
Cl- moves paracellularly (between cells) from lumen to interstitium, driven by the voltage gradient

IMG_0130.jpg
Explain this
analyse_media
Finding Sources
Reading File
Reading File
Here is a complete, structured explanation of everything in this image.
Renal Tubular Acidosis (RTA) - Full Breakdown
RTA is a group of disorders where the kidney fails to properly acidify the urine, leading to hyperchloremic normal anion gap metabolic acidosis. There are 3 types tested on USMLE: Type 1, Type 2, and Type 4 (Type 3 is not clinically distinct and is not tested).
The Big Picture First - How the Kidney Normally Handles Acid
- Proximal tubule (PCT): Reabsorbs ~85% of filtered HCO3-. Carbonic anhydrase (CA) inside the cell converts CO2 + H2O → H2CO3 → H+ + HCO3-. The H+ is secreted into the lumen via Na+/H+ exchanger, combines with luminal HCO3-, and the HCO3- is effectively reclaimed. Also produces NH3 → NH4+ (main way to excrete acid)
- Distal tubule / Collecting duct: Alpha-intercalated cells use H+-ATPase to secrete H+ into urine, regenerating new HCO3- that enters the blood. This is where urine pH is finally lowered to as low as 4.5
Failure at either site = RTA.
RTA Type 1 - Distal RTA
The Defect
Alpha-intercalated cells in the collecting duct cannot secrete H+
- No H+ secretion → no new HCO3- is generated → blood becomes acidotic
- The body chronically buffers this acid by dissolving bone calcium phosphate → releases HCO3- as a buffer, but also dumps Ca2+ and phosphate into urine
Key Features
| Feature | Value | Why |
|---|---|---|
| Urine pH | >5.5 (always) | The distal tubule cannot acidify urine at all - it stays alkaline |
| Serum K+ | ↓ (hypokalemia) | Failure to secrete H+ means the collecting duct compensates by secreting more K+ instead; also acidosis causes aldosterone-like effect |
| Serum HCO3- | Very low (can be <10) | Progressive wasting |
Causes (mnemonic: "AACOS")
- Amphotericin B toxicity (damages the apical H+ pump directly)
- Analgesic nephropathy
- Congenital anomalies / urinary tract obstruction
- Autoimmune diseases - esp. SLE, Sjogren's syndrome
Key Association
- Calcium phosphate kidney stones - because urine pH is always alkaline (>5.5), calcium phosphate precipitates easily (unlike uric acid or cystine stones which form in acidic urine). Also, bone buffering releases extra Ca2+ into urine. This is the only RTA that causes stones.
RTA Type 2 - Proximal RTA
The Defect
Proximal convoluted tubule (PCT) cannot reabsorb HCO3-
- Normally the PCT reabsorbs ~85% of filtered HCO3-. In Type 2, this mechanism (usually the Na+/H+ exchanger or carbonic anhydrase) is broken
- HCO3- floods into the urine → metabolic acidosis
- Twist: The distal tubule (alpha-intercalated cells) still WORKS. So the kidney CAN acidify urine... but only after plasma HCO3- drops low enough that there is less HCO3- being filtered and delivered distally
Key Features
| Feature | Value | Why |
|---|---|---|
| Urine pH | Biphasic | Early (when plasma HCO3- is normal): urine pH >5.5 because so much HCO3- is spilling into urine. Late (after plasma HCO3- has dropped below the resorptive threshold): urine pH <5.5 because less HCO3- is being delivered and the distal tubule can finally acidify. |
| Serum K+ | ↓ (hypokalemia) | HCO3- in the urine carries a negative charge that drives K+ secretion in the collecting duct |
| Serum HCO3- | Moderate reduction; stabilizes at a new set point |
Causes
- Fanconi syndrome - generalized PCT dysfunction (can't reabsorb glucose, phosphate, uric acid, amino acids, AND HCO3-). Can be caused by:
- Multiple myeloma (light chains toxic to PCT)
- Wilson's disease, galactosemia, cystinosis
- Heavy metals (lead, mercury)
- Carbonic anhydrase inhibitors (acetazolamide) - directly block HCO3- reabsorption
- Multiple myeloma alone
Key Association
- Hypophosphatemic rickets (in Fanconi syndrome) - because phosphate is also wasted in urine → bones don't mineralize properly → rickets in children
RTA Type 4 - Hyperkalemic RTA
The Defect
Hypoaldosteronism OR aldosterone resistance
This is the most common RTA in clinical practice. The mechanism is different from Types 1 and 2:
- No aldosterone effect → PCT produces less NH3 (ammonia)
- Less NH3 available in the tubule to buffer H+ → less NH4+ excreted
- H+ accumulates → metabolic acidosis
The hyperkalemia is ALSO part of the problem: high K+ suppresses NH3 synthesis in the PCT (K+ and NH4+ compete for the same transport), further reducing acid excretion.
Key Features
| Feature | Value | Why |
|---|---|---|
| Urine pH | Variable | The alpha-intercalated cells and distal H+ secretion still work, so urine CAN be acidified. The problem is failure of NH4+ buffering, not H+ secretion itself |
| Serum K+ | ↑ (hyperkalemia) | This is the defining feature - no aldosterone = no K+ wasting |
Causes - Two Categories
↓ Aldosterone PRODUCTION:
- Diabetic hyporeninism (most common cause overall - diabetics get hyporeninemic hypoaldosteronism)
- ACE inhibitors / ARBs (block angiotensin II → less aldosterone)
- NSAIDs (reduce renin → less angiotensin → less aldosterone)
- Heparin (directly inhibits adrenal aldosterone synthesis)
- Cyclosporine
- Adrenal insufficiency (Addison's disease)
Aldosterone RESISTANCE (receptor/post-receptor defect):
- K+-sparing diuretics (spironolactone blocks mineralocorticoid receptor; amiloride/triamterene block ENaC directly)
- TMP-SMX (trimethoprim blocks ENaC like amiloride)
- Nephropathy due to obstruction
Master Comparison Table
| Feature | Type 1 (Distal) | Type 2 (Proximal) | Type 4 (Hyperkalemic) |
|---|---|---|---|
| Site of defect | Distal collecting duct | Proximal tubule (PCT) | PCT / aldosterone axis |
| Defect | Can't secrete H+ | Can't reabsorb HCO3- | ↓ NH3 synthesis (no aldosterone) |
| Urine pH | Always >5.5 | Biphasic (<5.5 late, >5.5 early) | Variable |
| Serum K+ | ↓ Low | ↓ Low | ↑ HIGH |
| Serum HCO3- | Very low | Moderate | Mild reduction |
| Key cause | Ampho B, SLE | Fanconi, carbonic anhydrase inhibitors | Diabetic hyporeninism, ACE-i, K+-sparing diuretics |
| Association | Calcium phosphate stones | Hypophosphatemic rickets | None specific |
| Treatment | Oral HCO3- (large doses) | Oral HCO3- (but harder - causes more HCO3- wasting) + thiazides | Fludrocortisone, fix hyperkalemia, dietary K+ restriction |
USMLE High-Yield Points
- "Always alkaline urine + stones + hypokalemia" = Type 1 (Distal) RTA
- "Acetazolamide/Fanconi + rickets" = Type 2 (Proximal) RTA
- "Hyperkalemia + diabetic patient + ACE-i" = Type 4 RTA - most common RTA overall
- No RTA Type 3 on the exam
- All three types = normal anion gap (hyperchloremic) metabolic acidosis - the chloride rises to compensate for lost HCO3-
- The missing type is Type 3, which is now considered a variant of Type 1 and is not clinically relevant for boards
This is a shared conversation. Sign in to Orris to start your own chat.