Lytic lesion with hyperclacemia in hematological condition differentials
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
Loading Image
Finding Sources
I now have a thorough picture from multiple authoritative textbooks. Let me compile the comprehensive answer.
Lytic Lesions + Hypercalcemia in Hematological Conditions: Differentials
This combination defines a classic set of hematological malignancies. The mechanism in each involves either local osteolysis (LOH), PTHrP secretion, or ectopic 1,25(OH)₂D production - all leading to increased bone resorption and calcium release.
1. Multiple Myeloma (PRIMARY and most common)
The prototypical cause. Myeloma is a plasma cell neoplasm and is the most common hematological condition to produce both lytic lesions and hypercalcemia simultaneously.
Mechanism of bone destruction:
- Myeloma cells secrete MIP-1α (CCL3) and upregulate RANKL while simultaneously suppressing OPG - this shifts the RANKL/OPG ratio strongly toward osteoclast activation
- Myeloma cells also produce Wnt pathway inhibitors that block osteoblast function, creating a purely lytic pattern with no new bone formation
- Net result: massive uncoupled osteolysis, hypercalcemia, and pathologic fractures
- (Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 570; Goldman-Cecil Medicine, RANKL/OPG section)
Radiological features:
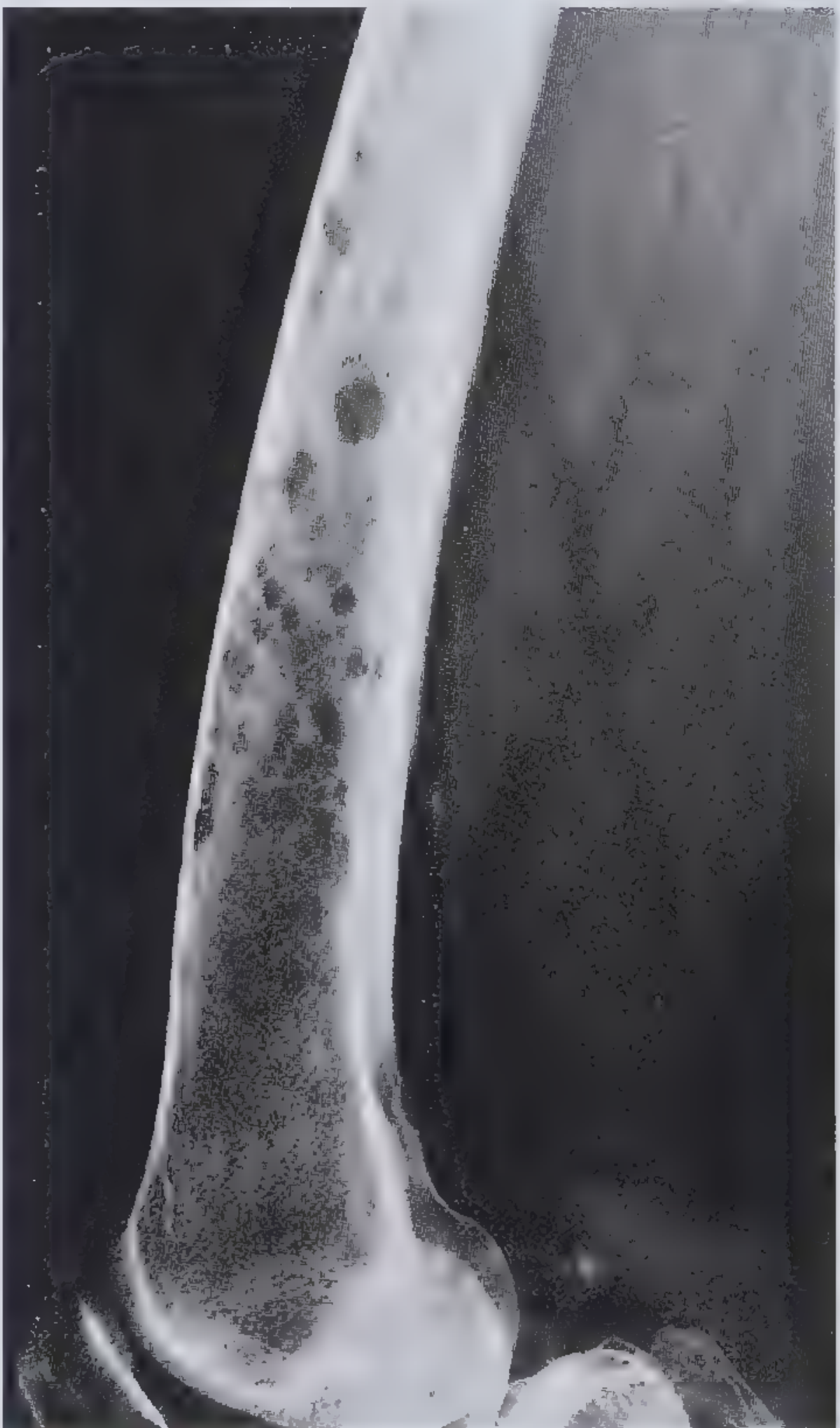
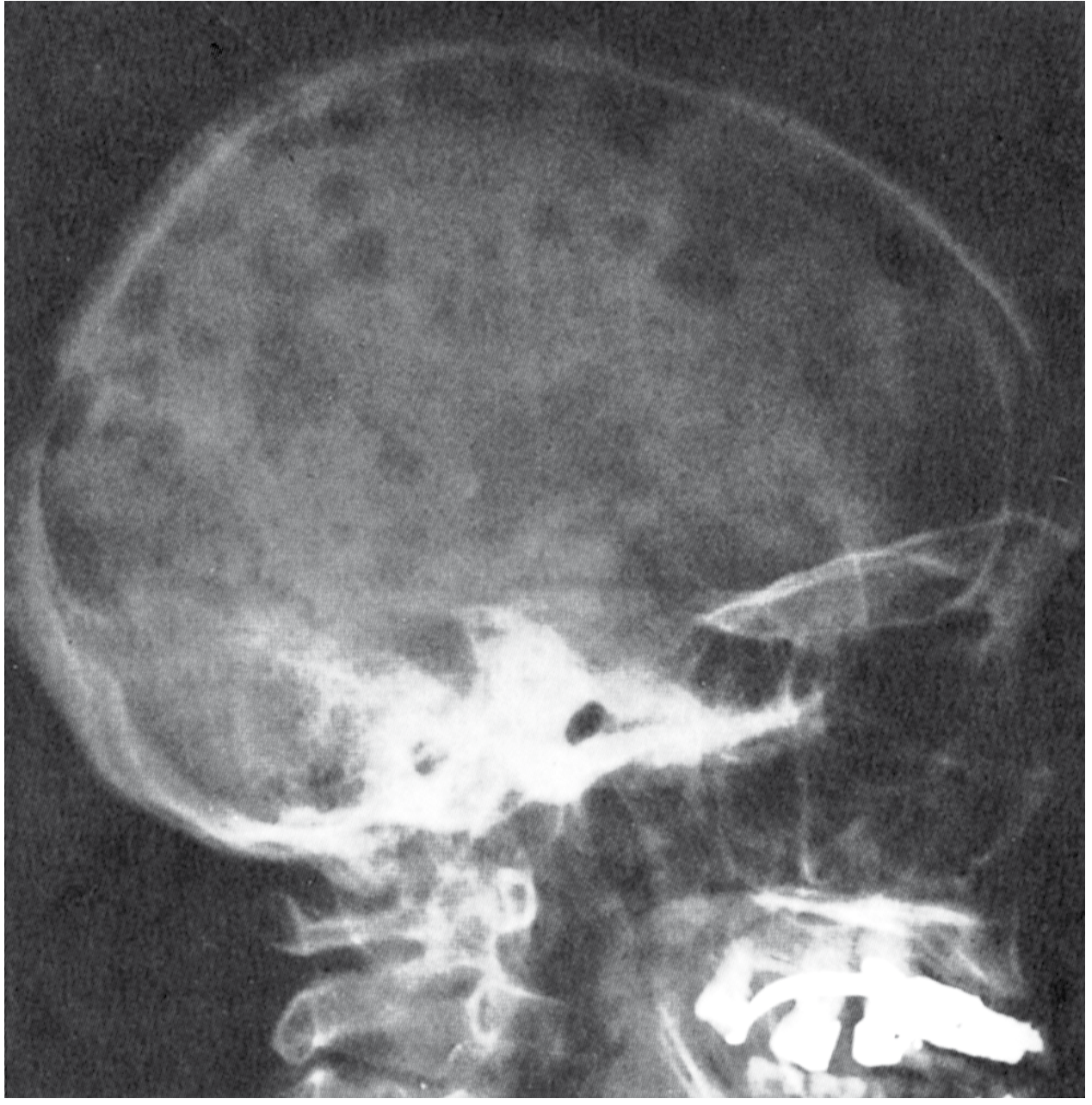
- "Punched-out" lytic defects, classically 1-4 cm, with no surrounding sclerosis (the absence of reactive new bone is characteristic)
- "Moth-eaten" appearance in long bones; calvaria lesions are highly characteristic
CRAB Criteria (myeloma-defining events):
| Letter | Manifestation |
|---|---|
| C | Hypercalcemia (>11 mg/dL or >1 mg/dL above upper normal) |
| R | Renal insufficiency (creatinine >2 mg/dL) |
| A | Anemia (Hb <10 g/dL) |
| B | Bone lesions (lytic lesions on skeletal survey/CT/PET) |
Additional myeloma-defining biomarkers: clonal bone marrow plasma cells >60%, serum FLC ratio >100, or >1 focal MRI lesion. (Comprehensive Clinical Nephrology 7th Ed; Quick Compendium of Clinical Pathology 5th Ed)
Bones affected (descending order): Vertebral column > ribs > skull > pelvis > femur > clavicle > scapula
Supporting features: M-protein on SPEP, Bence Jones proteinuria, rouleaux formation on blood smear, plasma cells >10% on bone marrow biopsy (CD138+, CD56+)
2. Adult T-Cell Leukemia/Lymphoma (ATLL) - HTLV-1 Associated
This is the second most important hematological condition to produce the lytic lesions + hypercalcemia combination.
- Driven by HTLV-1 (most prevalent in Japan and the Caribbean basin)
- Patients present with circulating disease, bone marrow involvement, hypercalcemia, lytic bone lesions, lymphadenopathy, hepatosplenomegaly, skin lesions, and opportunistic infections
- Pathognomonic: "flower cell" (CD4+, CD25+, CD2+, CD3+, CD5+, CD7-)
- 4 variants: acute (worst, median survival 6 months), lymphomatous, chronic, smoldering
- (Harrison's Principles of Internal Medicine 22E, 2025)
The hypercalcemia in ATLL is particularly aggressive - caused by PTHrP secretion by malignant T-cells and local osteoclast activation.
3. Non-Hodgkin Lymphoma (NHL)
Lymphoma causes hypercalcemia through two mechanisms:
| Mechanism | Details |
|---|---|
| Local osteolysis | Direct lytic bone involvement - all types of NHL can cause this |
| Ectopic 1,25(OH)₂D production | Malignant lymphocytes (especially in Hodgkin's and some NHL subtypes) convert 25(OH)D to active calcitriol via 1α-hydroxylase - this is PTHrP-independent |
The 1,25(OH)₂D pathway is particularly characteristic of lymphoma and distinguishes it from myeloma, where local osteolysis dominates.
(Frameworks for Internal Medicine; Brenner & Rector's The Kidney 2-vol)
4. Leukemias (less common, but recognized)
Certain leukemias can produce lytic lesions and hypercalcemia:
- AML (acute myeloid leukemia) - rare but reported
- CLL - uncommon, but B-cell infiltration of bone can produce lytic change
- Mechanism involves locally produced osteoclast-activating cytokines including PTHrP, IL-1, IL-6, and IL-8
- (Brenner & Rector's The Kidney - Malignancy section)
Summary Table: Hematological Differentials
| Condition | Lytic Lesions | Hypercalcemia | Key Mechanism | Distinguishing Feature |
|---|---|---|---|---|
| Multiple Myeloma | +++ | +++ | Local osteolysis (RANKL↑, OPG↓) | CRAB criteria, M-protein, BJ protein, punched-out lesions |
| ATLL (HTLV-1) | ++ | +++ | PTHrP + osteoclast activation | Flower cells, HTLV-1 serology, CD4+/CD25+/CD7- |
| NHL | ++ | ++ | Local osteolysis OR ectopic 1,25(OH)₂D | Elevated 1,25(OH)₂D (calcitriol), PTH suppressed |
| Hodgkin Lymphoma | + | + | Ectopic 1,25(OH)₂D | Reed-Sternberg cells, calcitriol-mediated |
| Leukemia (AML/CLL) | + | + | Osteoclast-activating cytokines | Peripheral blasts, bone marrow infiltration |
| Plasma cell leukemia | ++ | ++ | Same as myeloma | >20% circulating plasma cells, aggressive |
Key Differentiating Lab Workup
- SPEP/UPEP + immunofixation - M-protein = myeloma/plasmacytoma
- Serum free light chains - kappa/lambda ratio
- PTH - suppressed in all malignancy-related hypercalcemia
- PTHrP - elevated in ATLL, some NHL; low in myeloma
- 1,25(OH)₂D (calcitriol) - elevated in lymphoma-related hypercalcemia
- HTLV-1 serology - ATLL
- Bone marrow biopsy - plasma cell % (myeloma), T-cell immunophenotype (ATLL)
- Skeletal survey / Whole body low-dose CT - distribution and morphology of lytic lesions
Mnemonics:
- CRAB = the four cardinal myeloma manifestations (Calcium, Renal, Anemia, Bone)
- The absence of bone scan uptake (cold lesions) in myeloma vs. hot lesions in metastatic carcinoma is a classic radiological differentiator
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease; Harrison's Principles of Internal Medicine 22E (2025); Frameworks for Internal Medicine; Goldman-Cecil Medicine; Brenner & Rector's The Kidney 7th Ed
Tell me more differentials and perfect like hematologist minimum 5
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Excellent - I now have very rich, authoritative detail across all the additional differentials. Let me compile the full hematologist-level answer.
Lytic Lesions + Hypercalcemia in Hematological Conditions: Complete Differential (Hematologist Level)
This covers the full spectrum from plasma cell dyscrasias to histiocytic neoplasms, with mechanisms, distinguishing features, and clinical pearls at each level.
TIER 1 - Plasma Cell Dyscrasias (Most Likely)
1. Multiple Myeloma
(Already covered - see above for full CRAB criteria, morphology, and imaging)
The anchor diagnosis. Key reinforcement points for a hematologist:
- Bone scan is characteristically cold (purely lytic, no reactive new bone = no osteoblast activity = no uptake) - use skeletal survey or PET/CT/whole-body low-dose CT instead
- RANKL↑ / OPG↓ ratio is the core pathophysiologic driver
- IgG (52%) > IgA (21%) > light chain only (16%) > IgD (2%) > IgE (rare) - IgD and IgE myelomas have disproportionately high rates of plasma cell leukemia transformation
- Bones affected descending: vertebrae > ribs > skull > pelvis > femur > clavicle > scapula
- Robbins, Cotran & Kumar; Harrison's 22E (2025)
2. Solitary Plasmacytoma of Bone
A localized variant on the myeloma spectrum. Clinically important to distinguish because it is potentially curable.
| Feature | Detail |
|---|---|
| Definition | Single bony lytic mass of clonal plasma cells, no evidence of systemic myeloma |
| Bone marrow | <10% plasma cells at non-affected sites |
| M-protein | May be present, but without suppression of normal immunoglobulins |
| CRAB | Absent by definition (if CRAB present → reclassify as myeloma) |
| Hypercalcemia | Can occur, but far less common than in myeloma |
| Treatment | Radiation therapy (curative intent) - local control achieved in most |
| Key caveat | Two-thirds eventually progress to overt myeloma - lifelong surveillance is mandatory |
The hematologist must rule out occult systemic disease with PET-CT + bone marrow biopsy from a non-affected site before labeling it solitary.
(Henry's Clinical Dx; Goldman-Cecil; Robbins Cotran)
3. Plasma Cell Leukemia (PCL)
The most aggressive plasma cell dyscrasia - a hematological emergency.
- Diagnostic threshold: >5% plasma cells in peripheral blood differential and/or absolute plasma cell count ≥500/µL
- Primary PCL (60%): de novo presentation in leukemic phase - younger patients, more hepatosplenomegaly, lymphadenopathy, higher platelet count, fewer bone lesions, smaller M-protein, but better survival than secondary PCL
- Secondary PCL (40%): leukemic transformation of known myeloma - older patients, more bone disease, rapidly fatal
- Immunophenotype: unlike typical myeloma, CD56 is often absent in PCL
- IgD (12%) and IgE (25%) myelomas are over-represented among PCL transformations
- Hypercalcemia present due to same RANKL/osteoclast mechanism
- Treatment: aggressive induction (VDT-PACE or daratumumab-VRd) → ASCT if response achieved
- Goldman-Cecil Medicine; Harrison's 22E; Robbins Cotran
4. Nonsecretory Myeloma
- 3% of myeloma cases - no detectable M-protein in serum or urine by SPEP/UPEP/immunofixation
- Bone lesions and hypercalcemia are present and identical to secretory myeloma
- Diagnosis relies on bone marrow biopsy with immunoperoxidase/flow cytometry confirming clonal plasma cells
- Serum free light chain (sFLC) assay is abnormal in >60% - this is the key monitoring tool
- Pitfall: can be missed if sFLC not ordered; the clinician must have high suspicion when CRAB features present but standard protein studies are negative
- Goldman-Cecil Medicine
5. POEMS Syndrome (Osteosclerotic Myeloma)
A rare but clinically important plasma cell dyscrasia - the bone lesions here are osteosclerotic or mixed rather than purely lytic, but lytic lesions can occur.
| Component | Detail |
|---|---|
| P | Polyneuropathy (chronic inflammatory demyelinating - predominantly motor) |
| O | Organomegaly (liver, spleen, lymph nodes) |
| E | Endocrinopathy (hypogonadism, hypothyroidism, adrenal insufficiency, diabetes) |
| M | M-protein (almost always λ light chain) |
| S | Skin changes (hyperpigmentation, hypertrichosis, hemangiomas, clubbing) |
- Hypercalcemia and renal insufficiency are rare in POEMS (key differentiator from myeloma)
- Bone marrow usually has <5% plasma cells
- The diagnosis is confirmed by biopsy of a sclerotic lesion showing monoclonal plasma cells
- If lesions are limited: radiation therapy (>50% neuropathy improvement)
- If widespread: ASCT or systemic therapy
- Goldman-Cecil Medicine
TIER 2 - Lymphoid Malignancies
6. Adult T-Cell Leukemia/Lymphoma (ATLL) - HTLV-1 Driven
(From previous answer, expanded)
- The hematologist must ask about origin: Japan, Caribbean basin, West Africa, Middle East - HTLV-1 endemic regions
- Hypercalcemia in ATLL is mediated by PTHrP secreted by malignant T-cells, often dramatically elevated, can reach severe levels (>14 mg/dL)
- Lytic lesions + hypercalcemia + flower cells on smear = pathognomonic triad
- CD4+, CD25+, CD2+, CD3+, CD5+, CD7- (the CD7 loss is a key negative)
- FOXP3+ (resembles regulatory T-cells - contributes to profound immunosuppression)
- Skin involvement: erythroderma, nodules, papules (not just rash - actual leukemic skin infiltration)
- Opportunistic infections mirror AIDS because CD4 cells are dysfunctional despite being numerous
- Acute form: survival 6 months; treatment with zidovudine + interferon-α ± arsenic, mogamulizumab (anti-CCR4)
- Harrison's Principles of Internal Medicine 22E (2025)
7. Non-Hodgkin Lymphoma (NHL) with Bone Involvement
Two distinct subtypes of hypercalcemia mechanism within NHL:
A. Local Osteolysis (LOH) - diffuse large B-cell lymphoma, follicular lymphoma, Burkitt lymphoma
- Direct bone marrow infiltration → cytokine-mediated osteoclast activation (PTHrP, IL-1, IL-6, IL-8)
- Lytic lesions on imaging; PTH suppressed, PTHrP may be elevated
- Less commonly produces discrete punched-out defects (more permeative/infiltrative pattern)
B. Ectopic 1,25(OH)₂D (Calcitriol) Production - common in all lymphoma types
- Malignant lymphocytes express 1α-hydroxylase (same enzyme as granulomatous macrophages)
- Converts 25(OH)D → active 1,25(OH)₂D without PTH regulation
- Lab pattern: PTH suppressed ↓, PTHrP normal, serum 1,25(OH)₂D elevated ↑
- This is the most important distinguishing lab from myeloma (where calcitriol is NOT elevated)
- Can occur even without direct bone involvement
- All types of lymphoma can cause this syndrome
- (Brenner & Rector's The Kidney; Frameworks for Internal Medicine)
8. Hodgkin Lymphoma (HL)
- Lytic bone lesions and hypercalcemia occur but are less common than in NHL/myeloma
- Mechanism primarily ectopic 1,25(OH)₂D production (same as NHL above)
- Bone involvement in HL is usually part of stage IV disease
- Lytic lesions in HL can cause cord compression, pathologic fractures
- Key clinical point: HL is one of the few metastatic bone diseases that is potentially curable even with bone involvement - must not be mistaken for myeloma and written off as palliative
- (Harrison's 22E; Goldman-Cecil)
TIER 3 - Histiocytic Neoplasms
9. Langerhans Cell Histiocytosis (LCH)
Clonal proliferation of Langerhans cells - classified as a myeloid neoplasm in modern WHO taxonomy (driven by BRAF V600E in ~57% of cases).
| Disease Spectrum | Features |
|---|---|
| Eosinophilic granuloma (unifocal) | Single lytic bone lesion, usually asymptomatic |
| Hand-Schüller-Christian (multifocal unisystem) | Classic triad: exophthalmos + diabetes insipidus + skull lytic lesions; 15-40% of cases |
| Letterer-Siwe (multisystem) | Fulminant multiorgan disease; skin, bone, marrow, liver, spleen; often fatal without chemo |
| Multisystem with risk-organ involvement | Worst prognosis; liver/spleen/bone marrow ("risk organs") determine prognosis |
Bone lesions in LCH:
- "Punched-out" lytic lesions of the skull - can resemble myeloma radiologically
- CT shows sharp, well-defined lytic defect with beveled edges (geographic lysis)
- Preferential involvement of the calvaria, mandible, and temporal bone (causes aural polyp, conductive hearing loss, postauricular swelling)
- Hypercalcemia occurs but is less prominent than in myeloma
- Key differentiator: age (children/young adults), Birbeck granules on EM, CD1a+ / CD207 (Langerin)+ / S100+ on IHC
- BRAF V600E mutation - has therapeutic implications (vemurafenib responsive)
- (Goldman-Cecil Medicine; Robbins & Kumar Basic Pathology; Grainger & Allison's Diagnostic Radiology)
TIER 4 - Rare/Advanced Differentials (Complete the Hematologist's List)
10. Erdheim-Chester Disease (ECD)
- Non-Langerhans cell histiocytosis (CD68+, CD163+, CD1a-, S100-)
- BRAF V600E in ~54% of cases
- Bilateral and symmetrical long bone involvement (femora, tibiae, humeri) - the PET-CT pattern is distinctive (bilateral periosteal uptake)
- Lytic lesions less common than sclerotic lesions, but mixed patterns occur
- Hypercalcemia can occur
- Retroperitoneal fibrosis ("hairy kidney"), periaortic cuffing, diabetes insipidus, xanthelasma are hallmarks
- (Goldman-Cecil Medicine - described as a differential to LCH in the same section)
11. Hemophagocytic Lymphohistiocytosis (HLH) secondary to hematological malignancy
- Bone marrow infiltration by underlying lymphoma/leukemia triggers the syndrome
- Hypercalcemia via direct bony involvement + cytokine storm (IL-6, TNF-α)
- Context: ferritin >10,000, fever, splenomegaly, cytopenias, hypofibrinogenemia, elevated sCD25
Master Comparison Table (Hematologist Reference)
| Condition | Lytic Lesion | Hypercalcemia | Key Mechanism | Unique Lab/Feature | Bone Pattern |
|---|---|---|---|---|---|
| Multiple Myeloma | +++ | ++ | RANKL↑/OPG↓, MIP-1α | M-protein, BJ protein, CD138+ | Punched-out, NO sclerosis |
| Solitary Plasmacytoma | + (single) | +/- | Same as myeloma | Single lesion; 2/3 → myeloma | Single punched-out |
| Plasma Cell Leukemia | ++ | ++ | Same as myeloma | >5% plasma cells in blood, CD56- | Fewer lesions than MM |
| Nonsecretory Myeloma | +++ | ++ | Same as myeloma | Normal SPEP, sFLC assay key | Same as MM |
| POEMS | + (sclerotic) | Rare | - | λ-chain, neuropathy, VEGF↑ | Sclerotic not lytic |
| ATLL | ++ | +++ | PTHrP from T-cells | HTLV-1+, flower cells, CD4+/CD7- | Diffuse infiltrative |
| NHL | ++ | ++ | Osteolysis OR ectopic 1,25(OH)₂D | 1,25(OH)₂D elevated (not in MM) | Permeative |
| Hodgkin Lymphoma | + | + | Ectopic 1,25(OH)₂D | Reed-Sternberg, calcitriol↑ | Stage IV, variable |
| LCH | ++ | + | Cytokine-mediated osteolysis | CD1a+, Langerin+, BRAF V600E | Beveled-edge, geographic |
| ECD | +/- | +/- | Histiocyte infiltration | CD68+, CD1a-, bilateral long bone | Sclerotic > lytic |
Diagnostic Algorithm - Hematologist Approach
Lytic lesion(s) + Hypercalcemia
│
├── Check PTH → Suppressed in ALL malignancy-related causes
│
├── SPEP/UPEP + Immunofixation + sFLC ratio
│ ├── M-protein present → Plasma cell dyscrasia spectrum
│ │ ├── Multiple lesions + CRAB → MULTIPLE MYELOMA
│ │ ├── Single lesion, no systemic disease → SOLITARY PLASMACYTOMA
│ │ ├── >5% plasma cells in blood → PLASMA CELL LEUKEMIA
│ │ ├── No M-protein on SPEP but sFLC abnormal → NONSECRETORY MYELOMA
│ │ └── Sclerotic lesions + neuropathy + λ-chain → POEMS
│ │
│ └── No M-protein → Consider lymphoid or histiocytic cause
│ ├── Check PTHrP elevated? → ATLL, some NHL
│ ├── Check 1,25(OH)₂D elevated? → NHL, Hodgkin lymphoma
│ ├── HTLV-1 serology positive + flower cells → ATLL
│ ├── Lymph node biopsy / PET-CT → NHL / Hodgkin
│ └── Young patient + skull/temporal lytic lesions → LCH (CD1a, BRAF)
│
└── Bone marrow biopsy in all cases
Key Calcium Mechanism Summary for Exams & Boards
| Mechanism | Conditions | Lab Signature |
|---|---|---|
| Local osteolysis (RANKL/cytokines) | MM, PCL, ATLL, NHL, LCH | PTH↓, PTHrP normal, 1,25(OH)₂D normal |
| PTHrP-mediated osteolysis | ATLL, some NHL | PTH↓, PTHrP↑, 1,25(OH)₂D normal |
| Ectopic 1,25(OH)₂D | Hodgkin + NHL | PTH↓, PTHrP normal, 1,25(OH)₂D↑ |
Sources: Goldman-Cecil Medicine (International Edition); Harrison's Principles of Internal Medicine 22E (2025); Robbins, Cotran & Kumar Pathologic Basis of Disease; Robbins & Kumar Basic Pathology; Grainger & Allison's Diagnostic Radiology; Brenner & Rector's The Kidney 7th Ed; Frameworks for Internal Medicine; Henry's Clinical Diagnosis and Management by Laboratory Methods
If patient becomes allergic to l aspariginase then how to proceed
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Searching PubMed
"asparaginase" AND "hypersensitivity" AND "ALL" AND "management"
Searching PubMed
"asparaginase" AND "hypersensitivity" AND "Erwinia"
Finding Sources
Finding Sources
Reading Article
PMID: 35107320
Reading Article
PMID: 36795447
I now have comprehensive material. Here is the complete, hematologist-level answer.
L-Asparaginase Allergy: How to Proceed
This is a high-stakes clinical problem in ALL treatment because asparaginase is a non-negotiable component of ALL regimens - asparaginase exposure duration is one of the most important determinants of outcome. Losing it is associated with significantly worse event-free survival. The goal is to never abandon asparaginase therapy if at all possible - always switch formulation rather than stop.
Step 1 - Classify the Reaction First
Not all adverse reactions to asparaginase are the same. The clinical approach depends critically on the type of reaction.
| Reaction Type | Mechanism | Clinical Features | Action |
|---|---|---|---|
| Overt clinical hypersensitivity | IgE/IgG-mediated immune response to foreign bacterial protein | Urticaria, angioedema, bronchospasm, anaphylaxis during/after infusion | Stop current formulation → switch |
| Silent inactivation (subclinical) | Antibody-mediated enzyme inactivation WITHOUT clinical symptoms | No allergic signs, but asparaginase activity level undetectable | Clinically misleading - treatment failing silently |
| Grade 1-2 infusion reaction | Mild cytokine release or local reaction | Flushing, mild urticaria only | May attempt premedication first (see below), then reassess |
Key Pearl: Silent inactivation is more common than overt hypersensitivity and is associated with a negative clinical outcome in high-risk ALL because the enzyme appears to be given but is actually not working. Measuring serum asparaginase activity (SAA) is the only way to detect this.
Step 2 - Immediate Management of the Acute Reaction
- Stop the infusion immediately
- Administer:
- Epinephrine (for anaphylaxis: 0.01 mg/kg IM, max 0.5 mg)
- IV antihistamines (diphenhydramine)
- IV corticosteroids (hydrocortisone or methylprednisolone)
- IV fluids; oxygen; bronchodilators as needed
- Observe for minimum 1-2 hours post-reaction
- Document the reaction grade per CTCAE criteria (Grade 1-4)
- Check serum asparaginase activity level - if reaction occurred after a previous dose, retroactively check if prior doses had adequate activity
Step 3 - The Formulation Switch Strategy
This is the core of management. There are three distinct asparaginase preparations, derived from two different bacterial species with no immunological cross-reactivity between the two species:
The Three Formulations
| Formulation | Source | t½ | Dosing | Role |
|---|---|---|---|---|
| Native E. coli L-asparaginase (Elspar) | Escherichia coli | ~1 day | 6,000-10,000 IU every 3 days | Historical first-line (now rarely used as monotherapy) |
| Pegaspargase (PEG-asparaginase) (Oncaspar) | E. coli + PEG conjugate | 6-7 days | IM/IV every 14 days | Current first-line standard for ALL |
| Calaspargase pegol (Asparlas) | E. coli + different PEG linkage | >14 days | IV every 21 days | Newer, longer-acting; approved for ALL |
| Asparaginase Erwinia chrysanthemi (Erwinaze / JZP458/crisantaspase) | Erwinia chrysanthemi | ~16 hours (very short) | IM 3x/week or IV | For patients allergic to E. coli-derived formulations |
(Goodman & Gilman's Pharmacological Basis of Therapeutics; Harriet Lane Handbook 23rd Ed)
The Decision Tree
Patient develops hypersensitivity or silent inactivation to:
│
├── NATIVE E. COLI asparaginase
│ └── Switch to PEGASPARGASE (PEG-asparaginase)
│ (PEG coating reduces immunogenicity: <20% develop antibodies)
│
├── PEGASPARGASE (first-line) → allergic reaction
│ └── Switch to ERWINIA asparaginase (Erwinaze / JZP458)
│ (Completely different bacterial source = no cross-reactivity)
│ - Dose: 25,000 IU/m² IM 3× per week (Mon/Wed/Fri)
│ OR 25,000 IU/m² IV over 1 hour 3× per week
│ - Activity window is SHORT (t½ ~16h), hence the 3×/week dosing
│
├── ERWINIA asparaginase → shortage/unavailable
│ └── JZP458 (recombinant Erwinia-derived, produced in Pseudomonas)
│ - Approved 2021; closes the "asparaginase allergy gap"
│ - Produced recombinantly, not dependent on bacterial fermentation →
│ addresses supply shortage problem
│ [Blood, 2023 - PMID 36795447]
│
└── Allergic to ALL formulations (very rare)
└── Asparaginase discontinuation as LAST RESORT
- Intensify other components of regimen (consult protocol-specific guidance)
- Do NOT simply omit without expert hematology/oncology discussion
Step 4 - Why Cross-Reactivity Does NOT Occur Between Species
This is the pharmacological basis for the switch:
- Pegaspargase and native E. coli asparaginase: Both derived from E. coli - share the same protein epitopes. Antibodies raised against one will cross-react with the other. If pegaspargase causes hypersensitivity, switching to native E. coli asparaginase is not an option - both will be inactivated.
- Erwinia chrysanthemi asparaginase: Derived from a completely different organism with different protein structure and epitopes. Anti-E. coli asparaginase antibodies do not bind Erwinia enzyme. This is why the switch works.
Step 5 - Key Additional Considerations
Premedication Before Subsequent Doses
Some protocols premedicate with:
- Antihistamines (cetirizine or diphenhydramine)
- Acetaminophen
- Low-dose corticosteroids
However, premedication does not reliably prevent silent inactivation and may mask allergic symptoms without restoring enzyme activity.
Therapeutic Drug Monitoring (TDM) - Asparaginase Activity Levels
This is the most important monitoring tool in modern practice:
- Target serum asparaginase activity (SAA): ≥0.1 IU/mL at nadir (trough)
- Check SAA ~72 hours after E. coli preparations, ~7 days after pegaspargase
- If SAA <0.1 IU/mL (even without clinical reaction) = silent inactivation = switch formulation immediately
- (Future Oncology, Burke & Zalewska-Szewczyk, 2022 - PMID 35107320)
Managing Other Asparaginase Toxicities (Not Allergy)
These are separate toxicities requiring specific management (do NOT confuse with allergy - these may not require formulation switch alone):
| Toxicity | Mechanism | Management |
|---|---|---|
| Pancreatitis | Direct pancreatic damage | Hold asparaginase; if severe/necrotizing, may need permanent discontinuation; amylase/lipase monitoring |
| Coagulopathy/Thrombosis | ↓ antithrombin III, fibrinogen, protein C/S | Monitor coag profile; consider AT-III replacement; LMWH for thrombosis |
| Hemorrhage | ↓ clotting factors (fibrinogen, factors V, VII, VIII, X) | FFP if active bleeding; cryoprecipitate for fibrinogen <1 g/L |
| Hyperglycemia | ↓ insulin synthesis (protein synthesis inhibition) | Insulin sliding scale; monitor glucose closely |
| Hepatotoxicity | Protein synthesis inhibition → ↑ transaminases | Monitor LFTs; usually reversible |
| Hypoalbuminemia | ↓ albumin synthesis | Monitor albumin; affects pharmacokinetics of other drugs |
| Hyperammonemia | Ammonia release from asparagine hydrolysis | Rifaximin; lactulose; dietary protein restriction |
| Intracranial thrombosis/hemorrhage | AT-III ↓ + coagulopathy | Urgent MRI brain; anticoagulation for thrombosis; neurosurgery consult |
Summary Algorithm
L-Asparaginase allergy/silent inactivation confirmed
│
▼
Measure SAA → if <0.1 IU/mL = inactivation confirmed
│
▼
Identify source: E. coli-derived (native or PEG)?
│ │
Yes: SWITCH to Already on Erwinia?
ERWINIA asparaginase │
│ Yes: Use JZP458 (recombinant Erwinia)
▼ or asparaginase discontinuation
Continue ALL protocol as absolute last resort
with Erwinia dosing
(25,000 IU/m² 3×/week IM or IV)
│
▼
Monitor SAA 72h post-dose → target ≥0.1 IU/mL
Continue TDM throughout course
Bottom line for a hematologist: The most important principle is that asparaginase exposure must be maintained - it is a pillar of ALL cure rates. The E. coli to Erwinia switch is well-established, pharmacologically sound, and should be made promptly at the first sign of clinical allergy or silent inactivation confirmed by SAA monitoring. JZP458 (recombinant crisantaspase) is the newest option addressing Erwinia supply shortages.
Sources: Goodman & Gilman's Pharmacological Basis of Therapeutics; Harriet Lane Handbook 23rd Ed; Harrison's Principles of Internal Medicine 22E (2025); Burke & Zalewska-Szewczyk, Future Oncol 2022 [PMID 35107320]; Ribera, Blood 2023 [PMID 36795447]
Should i give peg asparagonase next time then
This is a critical clinical safety question - the answer depends entirely on which formulation the patient was originally on when they reacted.
The Core Principle: E. coli Cross-Reactivity
Pegaspargase (PEG-asparaginase) and native L-asparaginase are BOTH derived from E. coli.
They share the same underlying protein structure and antigenic epitopes. The PEG coating reduces immunogenicity - but it does not eliminate the cross-reactive protein core.
The Answer Depends on the Scenario
Scenario A: Patient reacted to native E. coli L-asparaginase
→ YES, pegaspargase is an appropriate next step
- The PEG conjugation significantly reduces immunogenicity
- Less than 20% of patients develop antibodies to pegaspargase vs. much higher rates with native E. coli form
- Goodman & Gilman explicitly states: "In hypersensitive patients [to native E. coli], pegaspargase is a safe and effective alternative"
- This switch is well-supported and widely practiced
Scenario B: Patient reacted to pegaspargase itself
→ NO - do NOT give pegaspargase again. This is contraindicated.
This is the more common and more dangerous scenario in modern practice because pegaspargase is now first-line in most ALL protocols.
- The antibodies raised against pegaspargase will still recognize and neutralize any E. coli-derived asparaginase, including calaspargase pegol
- Giving pegaspargase again risks anaphylaxis or guaranteed silent inactivation
- The only correct move here is Erwinia asparaginase (Erwinaze / JZP458/crisantaspase) - completely different bacterial source, no cross-reactivity
Decision Table
| Patient reacted to | Can you give Pegaspargase? | Correct next step |
|---|---|---|
| Native E. coli asparaginase | YES | Pegaspargase |
| Pegaspargase | NO - contraindicated | Erwinia asparaginase |
| Calaspargase pegol | NO | Erwinia asparaginase |
| Erwinia asparaginase | N/A | JZP458 (recombinant Erwinia) |
Why This Distinction Matters Clinically
Modern ALL protocols (COG AALL0434, BFM, UKALL) now use pegaspargase as the default first-line formulation. So when a patient on a modern ALL protocol develops allergy, they are almost always reacting to pegaspargase - and the answer is Erwinia, not more PEG.
The scenario where you would switch to pegaspargase is increasingly uncommon, seen mainly when:
- The patient was started on native E. coli asparaginase (older protocol, resource-limited setting)
- The allergy occurred during the older native formulation phase before PEG was introduced
One More Critical Point: Silent Inactivation
Even if you switch from native E. coli → pegaspargase and the patient has no clinical allergic reaction, you must check serum asparaginase activity (SAA) at nadir (≥0.1 IU/mL target).
Pre-formed anti-E. coli antibodies can silently inactivate pegaspargase with zero clinical symptoms. The patient appears to be receiving therapy but the drug is doing nothing - a dangerous situation that is only caught by TDM.
Bottom line: If your patient reacted to native E. coli asparaginase → pegaspargase is appropriate. If they reacted to pegaspargase → pegaspargase is contraindicated; use Erwinia asparaginase. Always monitor SAA regardless of which formulation you use next.
This is a shared conversation. Sign in to Orris to start your own chat.