Congenital adrenal hyperplasia
Reading File
Finding Sources
Searching PubMed
"congenital adrenal hyperplasia"[MeSH Terms]
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
Reading File
I now have comprehensive content from authoritative textbooks. Let me compile the full, detailed answer.
Congenital Adrenal Hyperplasia (CAH)
Overview
Congenital adrenal hyperplasia is a group of autosomal recessive disorders caused by enzyme defects in the cortisol biosynthetic pathway. The fundamental defect is inadequate cortisol synthesis, which removes negative feedback on the hypothalamus and pituitary, causing elevated CRH and ACTH. The resulting chronic ACTH excess drives adrenal hyperplasia and accumulation of steroid precursors proximal to the enzymatic block - many of which are shunted into androgen synthesis.
- Goldman-Cecil Medicine, p. 2515
Steroidogenesis Pathway
This is the map of the pathway - understanding it is key to understanding every form of CAH:
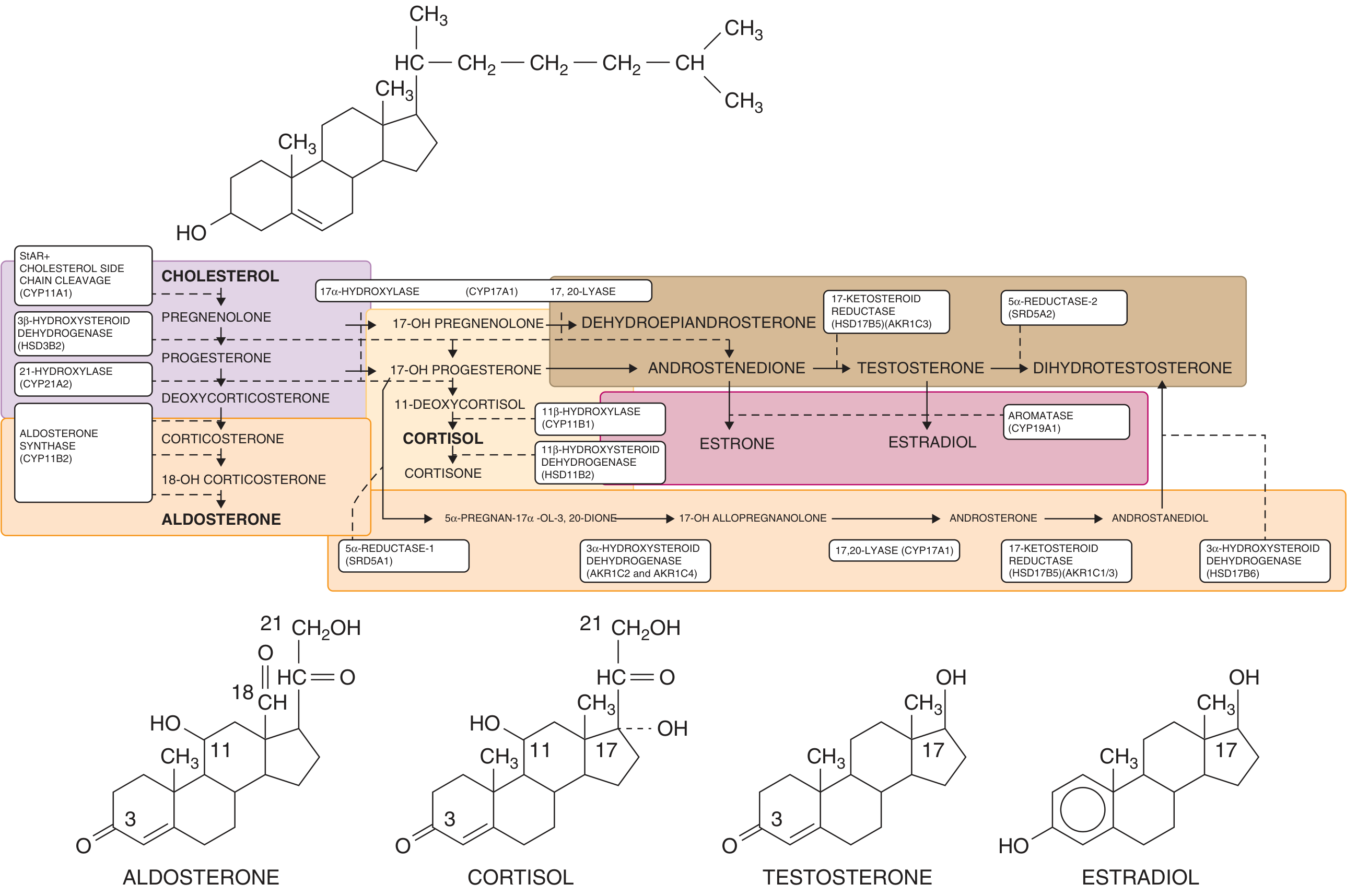
Steroidogenesis: cholesterol to aldosterone, cortisol, testosterone, and estradiol. Enzyme blocks at each step define the different forms of CAH. - Goldman-Cecil Medicine
Enzyme Defects and Forms of CAH
A defect in any of five enzymes can cause CAH. All are transmitted in an autosomal recessive pattern:
| Enzyme | Gene | Key Features |
|---|---|---|
| 21-Hydroxylase | CYP21A2 | 95% of all CAH; salt-wasting or simple virilizing |
| 11β-Hydroxylase | CYP11B1 | ~5%; virilization + hypertension (DOC accumulation) |
| 3β-Hydroxysteroid dehydrogenase | HSD3B2 | Rare; affects adrenal + gonads |
| 17α-Hydroxylase | CYP17A1 | Rare; hypertension, sexual infantilism |
| Cholesterol side-chain cleavage (StAR) | StAR/CYP11A1 | Congenital lipoid adrenal hyperplasia; severe adrenal insufficiency |
- Campbell-Walsh Urology, p. 1291
21-Hydroxylase Deficiency (Most Common Form)
Epidemiology
- Responsible for ~95% of all CAH cases
- Incidence: 1 in 5,000-15,000 in the US and Europe
- Highest incidence: 1 in 490 in the Yupik Alaskan Eskimo population
- Gene: CYP21A2 on chromosome 6p21.3, within the HLA complex
Genetics
- The CYP21A2 gene sits adjacent to a nearly identical pseudogene (CYP21PA1), which is 98% homologous but encodes no functional protein
- During meiosis, gene conversion transfers inactivating segments from the pseudogene to the functional gene
- ~10 mutations account for 90-95% of alleles; over 200 different CYP21 mutations have been described
- Campbell-Walsh Urology, p. 1291
Clinical Classification (Three Forms)
1. Salt-Wasting (75% of classic CAH)
- Both aldosterone AND cortisol deficient
- Life-threatening salt-wasting crisis in the neonatal period: hyponatremia, hyperkalemia, hypotension, shock
- Virilization of females at birth
- Males often diagnosed only after salt-wasting crisis (no obvious genital abniguity)
2. Simple Virilizing (25% of classic CAH)
- Cortisol deficient; aldosterone synthesis preserved
- Virilization without salt wasting
- Female: ambiguous genitalia at birth
- Male: precocious puberty
3. Non-Classic (Attenuated)
- Mild enzyme impairment; no virilization or salt wasting at birth
- Presents at or after puberty with hirsutism, menstrual irregularity, acne, reduced fertility in females
- Associated with family history of hirsutism
- Goldman-Cecil Medicine, p. 2550
Clinical Manifestations
In Females (46,XX)
- Classic forms cause virilization beginning at 10 weeks' gestation (formation of external genitalia)
- Clitoromegaly and labial fusion are present at birth in nearly all classic cases
- Vagina and urethra open into a common urogenital sinus
- In severe cases, genitalia may appear as a hypospadiac penis with cryptorchidism
- Prader staging (I-V) classifies the degree of virilization
- Müllerian structures (uterus, tubes) are normal - ovaries develop normally
- Campbell-Walsh Urology, p. 1291
In Males (46,XY)
- External genitalia appear normal at birth (no obvious abnormality)
- Salt-wasting crisis at 1-3 weeks of life is often the presenting event - a potentially lethal adrenal emergency
- Simple virilizing form: precocious puberty, rapid linear growth but early epiphyseal fusion causing short adult stature
- Testicular adrenal rest tumors (TARTs): ectopic adrenal tissue in the testes; can cause gonadal damage and infertility if not controlled
Both Sexes
- Advanced bone age and accelerated growth in childhood
- Short adult stature (due to premature epiphyseal fusion)
- Risk of adrenal insufficiency under stress (illness, surgery)
11β-Hydroxylase Deficiency
- Second most common form (~5%)
- Deficiency of CYP11B1 blocks conversion of 11-deoxycortisol to cortisol and 11-deoxycorticosterone (DOC) to corticosterone
- DOC accumulation causes sodium retention and hypertension (a distinguishing feature from 21-hydroxylase deficiency, which causes salt wasting)
- Virilization occurs in females similarly to 21-hydroxylase deficiency
- Androgen excess pathway is the same (precursors shunted to androgens)
Diagnosis
Neonatal Screening
- All 50 US states and >40 countries screen newborns for CAH
- Measures 17-hydroxyprogesterone (17-OHP) on dried blood spot (elevated in 21-hydroxylase deficiency)
- Has reduced time to diagnosis and mortality, especially in males with the salt-wasting form
Hormonal Evaluation
| Test | Finding in 21-OH Deficiency |
|---|---|
| 17-OHP (basal or ACTH-stimulated) | Markedly elevated |
| Cortisol | Low |
| Aldosterone | Low (salt-wasting form) |
| Plasma renin activity | Elevated (salt-wasting) |
| ACTH | Elevated |
| Androgens (testosterone, DHEA-S, androstenedione) | Elevated |
| Electrolytes | Hyponatremia, hyperkalemia (salt-wasting) |
- ACTH (cosyntropin) stimulation test with 17-OHP measurement is the gold standard for diagnosis of non-classic CAH
- Genetic testing confirms the specific CYP21A2 mutation
Karyotype
- Mandatory for any infant with ambiguous genitalia
Management
Glucocorticoid Replacement
- Hydrocortisone is the preferred agent in children (15-20 mg/m²/day in 2-3 divided doses)
- Suppresses ACTH, reduces adrenal androgen overproduction, prevents virilization progression
- In adults: hydrocortisone or prednisone
- Dose must be increased during illness, surgery, or physiological stress ("sick day rules")
Mineralocorticoid Replacement (Salt-Wasting Form)
- Fludrocortisone (0.05-0.2 mg/day) to replace aldosterone
- Sodium supplementation in infants (1-2 g/day)
Monitoring
- Growth, bone age, blood pressure
- 17-OHP, androstenedione, testosterone, renin levels
- Avoid over-treatment (causes iatrogenic Cushing syndrome and growth suppression)
Surgical Management
- Feminizing genitoplasty in severely virilized females: vaginoplasty and clitoroplasty
- Timing and extent remains controversial; trend toward deferring to adolescence or adulthood when possible (patient autonomy)
Prenatal Treatment (Investigational/Controversial)
- Dexamethasone (crosses placenta) given to the mother from early first trimester can reduce virilization of an affected female fetus
- Requires early diagnosis via chorionic villus sampling; risk-benefit ratio is debated
Adrenal Crisis Management
- IV hydrocortisone (50-100 mg/m² bolus) + IV saline/dextrose
- A medical emergency requiring immediate treatment
Other Rarer Forms
Congenital Lipoid Adrenal Hyperplasia (StAR Deficiency)
- Most severe form; impairs the first step (cholesterol → pregnenolone)
- Adrenal glands become large and lipid-laden
- 46,XY individuals have female or ambiguous external genitalia
- Present in neonatal period with severe adrenal insufficiency, salt wasting, hyponatremia, hyperkalemia, metabolic acidosis
- Common in Japanese, Korean, and Palestinian populations
- Campbell-Walsh Urology, p. 1292
3β-HSD Deficiency
- Affects both adrenal and gonads (common early pathway)
- Both sexes: salt-wasting and adrenal insufficiency
- Males: undervirilization (decreased testosterone); females: mild virilization (DHEA elevation)
17α-Hydroxylase/17,20-Lyase Deficiency
- Sex steroid synthesis impaired in both adrenal and gonads
- Result: sexual infantilism, primary amenorrhea in 46,XX; ambiguous/female genitalia in 46,XY
- Hypertension and hypokalemia from DOC and corticosterone accumulation
Key Points Summary
- CAH = autosomal recessive enzyme defect → cortisol deficiency → ACTH excess → adrenal hyperplasia + precursor accumulation → androgen excess
- 21-hydroxylase deficiency (CYP21A2) = 95% of cases; three forms: salt-wasting, simple virilizing, non-classic
- Salt-wasting crisis in neonates is a medical emergency
- Neonatal screening using 17-OHP is standard worldwide
- Treatment = lifelong glucocorticoid ± mineralocorticoid replacement
- 46,XX females with classic CAH have normal internal (Müllerian) structures but virilized external genitalia
- Males with classic CAH have normal external genitalia but may present with adrenal crisis
Sources: Goldman-Cecil Medicine (pp. 2515-2516); Campbell-Walsh Urology (pp. 1291-1295)
This is a shared conversation. Sign in to Orris to start your own chat.